
D-A 型二噻吩并磷杂茂衍生物分子的双光子吸收性质
2014-03-20 08:15:26丁红娟王传奎
原子与分子物理学报 2014年4期
丁红娟,王传奎
(山东师范大学物理与电子科学学院,济南250014)
1 引 言
双光子吸收 (TPA)是指分子从基态向高能电子态激发时同时吸收两个光子的过程.1931年,Göppert-Mayer在理论上预测了双光子吸收的可能性.1961年激光问世后才由Kaiser等首先从实验上证实了双光子吸收过程的存在[1].目前,具有大的双光子吸收截面的材料在三维光信息存储[2]、光 学 微 加 工[3]、光 限 幅[4]、光 动 力 学 治疗[5]和双光子荧光显微 (TPEP)成像[6]等许多领域展示出良好的应用前景.因此设计和合成具有功能性的双光子吸收材料成为理论和实验工作者非常感兴趣的课题.为了改善分子的双光子吸收截面值,人们开展了广泛的结构-性质关系的研究[7-10].已经报道的双光子吸收分子类型有二维的偶极分子 (D-π-A)和四极矩分子 (A-π-A,Dπ-D)以及多极的星形分子.已有的研究结果表明分子的双光子吸收截面与分子的平面性、共轭长度、π中心部分的特性、官能团的推拉电子能力、分子的对称性和分子的维度有关.此外,溶剂效应对分子的双光子吸收截面也有重要影响[11].
迄今,杂环已被大量用于分子的设计,而其中将磷原子引入有机分子材料引起了大家的关注[12-14],磷原子的多种反应活性以及电子性质为新型材料的发展提供了可能.而对于五元环磷杂茂系统并未得到广泛的研究.由于磷杂茂独特的锥形结构,阻碍了磷原子上的孤对电子和π共轭体系的相互作用,导致了孤对电子很小的离域性.通过磷原子的氧化和金属配合,改变磷原子的氧化态,可以方便的调控含磷共轭体系的电子结构.最近,Baumgartner等[15]合成了一系列以二噻吩磷杂茂为中心的不对称功能TPA 分子 (分别记为DTP0、DTP1和DTP2,见图1),并测量了它们的单光子和双光子吸收特性.DTP0 分子中心磷原子分别氧化为DTP1分子,金属配合为DTP2分子.三个分子都具有给体 (D)二苯胺基-NPh2和受体 (A)三氟代甲基-CF3,属于典型的D-A型偶极分子.实验结果表明三个分子在生物波长范围内都具有可观的双光子吸收截面值,并且无论中心的磷原子氧化和配合,该系列分子双光子吸收特性几乎不变.为了更加深入的了解这些分子的光吸收机制,我们在杂化的密度泛函 (DFT)理论水平上,利用响应理论方法计算了分子的线性吸收和双光子吸收性质.此外,由于实验是在溶剂环境下测量的,我们还考虑了溶剂效应,采用的是PCM 模型.
2 理论方法
我们采用HF 方法对分子的几何结构进行了全优化,选用赝势基组LANL2DZ 作为Au 原子的基组,其它原子选用基组6-31G 基组.该计算工作在Gaussian09程序包[16]上完成.然后采用响应理论方法在DFT/B3LYP水平上分别计算了分子的单光子和双光子吸收性质[7].其中,Au原子的基组采用lanl2dz_ecp基组,其他原子使用6-31G 基组.该计算工作在Dalton2011程序包[17]上完成.我们还考虑了溶剂效应对分子光学性质的影响,溶剂模型采用的是PCM 模型.
3 结果与讨论
3.1 分子结构
在HF 水平上优化了分子的几何结构.表1给出了优化后几何结构的一些键长和二面角情况.从表1可以看出对于金属配合的DTP2分子,两端的苯环相对于π 中心具有更大的扭转角.表1还给出了共轭方向上的一些单双键的键长以及键长的变化 (BLA)值.结果表明,DTP2 分子的BLA 值最大,为0.137Å,而DTP0 和DTP1 分子的键长相差很小,且BLA 值相同,都为0.125 Å.这说明了中心磷原子的金属配合引起了几何结构的较大变化,而磷原子的氧化对共轭体系几何结构的影响很小.

图1 分子结构示意图Fig.1 Schematic structures of molecules
此外,我们还在DFT/B3LYP 水平上优化了分子的几何结构,发现得到的结构具有更好的平面性.并且相对于HF 方法,采用DFT 优化得到的结构来计算分子的光学性质与实验结果有较大的差距.这主要因为采用DFT 方法优化的结构往往会低估共轭有机分子的键长变化,这和已有的研究结果[18]是一致的.因此对于该分子我们采用HF方法优化得到的结构来进行接下来的计算.
3.2 单光子吸收
在HF方法优化得到的结构的基础上,我们在DFT/B3LYP水平上采用线性响应理论方法计算了分子DTP0、DTP1和DTP2的单光子吸收性质.表2给出了气相下分子最低六个激发态的单光子吸收波长和振子强度.计算结果表明分子DTP0、DTP1和DTP2的最大单光子吸收态都出现在第一激发态,分别位于506.55、545.78 和528.44nm 处,这和实验上在DCM 溶剂中测得的结果分别为474、489 和479nm 是定性一致的.计算得到的DTP1和DTP2两个分子的单光子吸收与DTP0 分子相比都发生了一定程度的红移,但是红移程度不大 (<40nm).

表1 优化的几何结构的键长 (Å)和二面角 (C°)Table 1 Selective bond lengths(Å)and dihedral angles(C°)of the optimized geometries
进一步我们还计算了分子轨道的性质,分子DTP0、DTP1和DTP2 第一激发态主要来源于HOMO→LUMO 轨道的跃迁,所占比例分别为89.7%、90.4%和98.6%.分子DTP0、DTP1和DTP2的能隙大小分别为2.71、2.53和2.62eV.结果表明磷原子的氧化和金属络合使得能隙减小,从而导致了分子单光子吸收峰的红移.这主要是由于环外的P-R 键和π共轭体系的相互作用使得分子呈现出一些σ*–π*的超共轭特性,从而显著降低了LUMO 能级.
3.3 双光子吸收
在HF方法优化得到的结构的基础上,我们采用响应理论方法计算了气相下分子的双光子吸收性质.表3列出了DTP0、DTP1和DTP2分子最低六个激发态的双光子吸收波长和双光子吸收截面.理论计算结果表明在低能量范围内,分子的最大双光子吸收都发生在第二激发态,其最大的双光子吸收截面分别为2529、2372 和1902 GM (GM=10-50cm4s/photon),对应的双光子吸收波长位于817.68、854.08和810.85nm 处.与之前我们研究的DA 型分子相比[7],这三个分子都具有较大的光谱展宽和显著的双光子吸收截面,表明了三个分子都是较好的双光子吸收发色团.此外,我们可以看到分子都有两个较大的双光子吸收态,分别位于800nm 和1000nm 附近,并且DTP0、DTP1和DTP2分子的双光子吸收峰值都比较接近,这表明了DTP0分子中心的磷原子无论是被氧化还是金属配合,对其双光子吸收截面的影响都不大,这和实验结果是一致的.我们需要说明的是,理论计算的结果和实验结果有一定的差距,除了我们采用的近似方法外,理论和实验结果出现偏差主要由以下原因造成:理论值是与在静态条件下的共振双光子吸收有关的,而实验测量不容易达到共振条件;分子的光学性质受光和分子的相互作用的影响,实验测量给出的是动态双光子吸收截面;此外,在计算中没有考虑溶剂效应和分子的振动,这些都会对计算结果产生影响.

表2 气相中分子最低六个激发态的单光子吸收波长λop(nm)和振子强度δop (a.u.)Table 2 The OPA wavelengthsλop (nm)and the oscillator strengthsδop (a.u.)of the lowest six excited states for the compounds in gas phase
3.4 电荷转移过程
我们知道对于有机共轭分子电荷转移 (CT)态对分子的光学性质起到非常重要的作用.当分子从基态激发到CT 态的过程中,分子内的电荷会重新分布.我们计算了分子从基态跃迁到电荷转移态S1的过程中分子末端基团的电荷变化 (见表4).从表4可以看出在分子激发过程中,二苯胺基给出电子,表现为给体 (D),而三氟代甲基得到电子,表现为受体 (A).可见,电子从二苯胺基向三氟代甲基方向转移,DTP0、DTP1 和DTP2分子都是典型的D-A 型偶极分子.并且在从基态激发到电荷转移态的过程中,由于正负电荷的分离导致分子的极性显著增大.
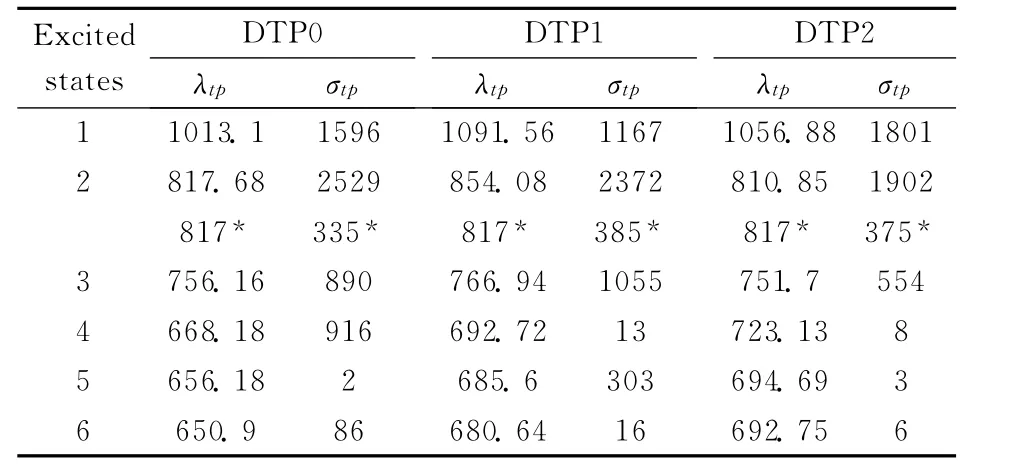
表3 气相中分子最低六个激发态的双光子吸收波长λtp(nm)和双光子吸收截面σtp (GM=10-50cm4s/photon)Table 3 The TPA wavelengthsλtp (nm)and the TPA cross sectionsσtp (GM=10-50cm4s/photon)of the lowest six excited states for the compounds in gas phase

表4 末端基团激发态S1 和基态S0 的电荷差异Table 4 The net charge differences between the first excited state S1and the ground state S0of terminal groups
3.5 溶剂效应
由于实验测量是在溶液中进行的,因此我们还考虑了溶剂对分子光学性质的影响.我们理论计算了在不同溶剂甲苯、氯仿、THF、DCM 和乙醇下DTP1分子的光学性质 (见表5),包括最大单光子吸收处的波长、单光子吸收强度、前线分子轨道能量和能隙、最大的双光子吸收处的波长和双光子吸收截面.此外,静电介电常数和光频介电常数也被考虑进来.计算结果表明:与气相下的结果相比,考虑溶剂效应时DTP1分子的单光子吸收峰发生了不同程度的蓝移,并且蓝移的程度和溶剂极性成非线性的关系.观察分子的前线轨道能量可以看到,与气相下的结果相比,HOMO 轨道和LUMO 轨道能级都有不同程度的升高,并且LUMO 轨道有更大程度的上升,这是由于处在CT 态的分子具有更大的极性.这导致了能隙的增宽,进而造成单光子吸收的蓝移.另外,单光子吸收的强度也随溶剂极性呈现一个非线性的变化趋势,并且更加依赖于光频介电常数.对于在不同溶剂下DTP1分子的双光子吸收情况,我们可以看到与气相下相比,分子的最大双光子吸收截面都有不同程度的增大,和溶剂极性也是呈现出非线性的关系.与气相下的结果相比,当考虑溶剂效应时所得到的理论计算结果与实验结果更加符合.可见当我们研究分子的光学性质时,溶剂效应是非常重要的因素.
4 结 论
利用响应理论方法在B3LYP 水平上计算了一系列2,6-donor-accepter二噻吩并磷杂茂衍生物分子的单、双光子吸收性质.此外,还采用PCM 模型研究了溶剂效应对分子光吸收性质的影响,结果表明分子发生了溶剂化蓝移,考虑溶剂效应得到的理论计算结果和实验结果更加符合,并且随溶剂极性变化,分子的单、双光子吸收表现出非线性行为.在生物波长范围内,该系列分子都具有显著的双光子吸收截面,是很好的双光子吸收材料.并且分子中心磷杂茂的磷原子无论被氧化还是金属配合,其双光子吸收截面几乎保持不变.这种特性可以被用来设计合成具有特殊用途的分子,可以改变分子的末端部分而保持其良好的双光子吸收特性.
表5 DTP1分子在不同溶剂下的光学性质,包括最大单光子吸收处的波长(nm)、单光子吸收强度x (a.u.)、前线分子轨道能量EH/L (eV)和能隙ΔEL-H (eV),以及最大的双光子吸收处的波长(nm)和双光子吸收截面(GM)Table 5 The optical properties in different solvents for DTP1,including the maximum OPA wave lengths(nm),the oscillator strengths(a.u.),the frontier orbital energies EH/L (eV)and energy gapsΔEL-H (eV),as well as TPA wave lengths(nm)and cross sections(GM)
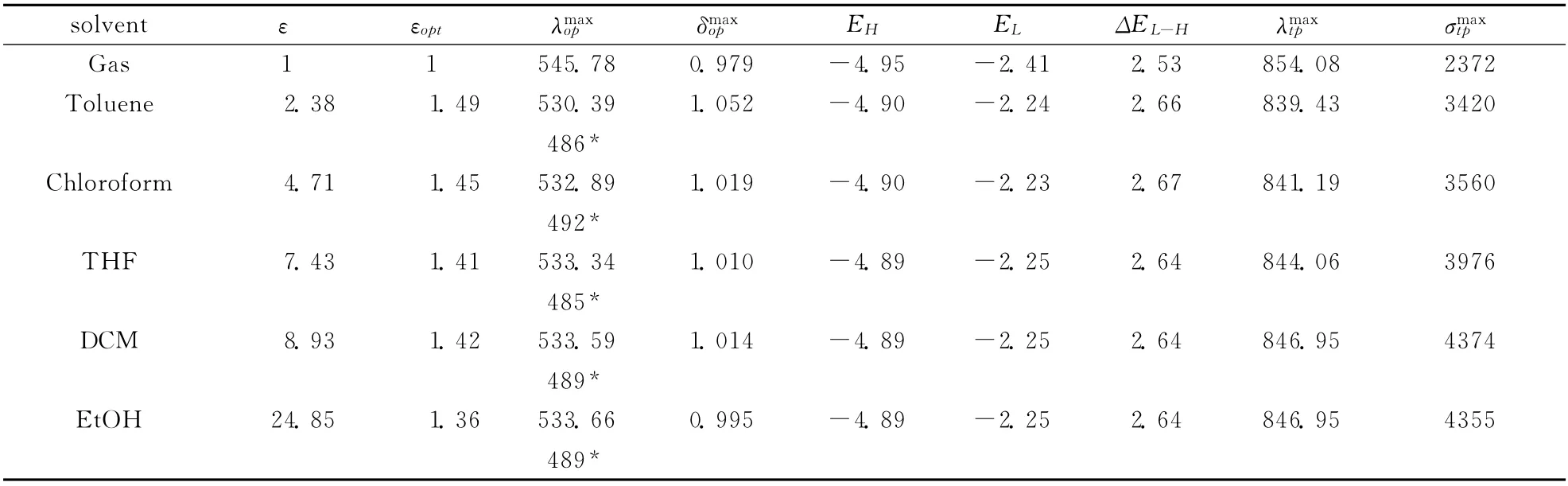
表5 DTP1分子在不同溶剂下的光学性质,包括最大单光子吸收处的波长(nm)、单光子吸收强度x (a.u.)、前线分子轨道能量EH/L (eV)和能隙ΔEL-H (eV),以及最大的双光子吸收处的波长(nm)和双光子吸收截面(GM)Table 5 The optical properties in different solvents for DTP1,including the maximum OPA wave lengths(nm),the oscillator strengths(a.u.),the frontier orbital energies EH/L (eV)and energy gapsΔEL-H (eV),as well as TPA wave lengths(nm)and cross sections(GM)
*Experimental data in different solvents taken from Ref.[15]
solvent ε εopt λmax op δmax op EH EL ΔEL-H λmax tp σmax t p Gas 1 1 545.78 0.979 -4.95 -2.41 2.53 854.08 237 2 Toluene 2.38 1.49 530.39 1.052 -4.90 -2.24 2.66 839.43 3420 486*Chloroform 4.71 1.45 532.89 1.019 -4.90 -2.23 2.67 841.19 3560 492*THF 7.43 1.41 533.34 1.010 -4.89 -2.25 2.64 844.06 3976 485*DCM 8.93 1.42 533.59 1.014 -4.89 -2.25 2.64 846.95 4374 489*EtOH 24.85 1.36 533.66 0.995 -4.89 -2.25 2.64 846.95 4355 489*
[1] Kaiser W,Garrett C G B.Two-photon excitation in CaF2:Eu2+[J].Phys.Rev.Lett.,1961,7(6):29.
[2] Ogawa K,Kobuke Y.Design of two-photon absorbing materials for molecular optical memory and photodynamic therapy[J].Org.Biomol.Chem.,2009,7:2241.
[3] Lee K S,Kim R H,Yang D Y,et al.Advances in 3Dnano/microfabrication using two-photon initiated polymerization[J].Prog.Polym.Sci.,2008,33(6):631.
[4] Four M,Riehl D,Mongin O,et al.A novel ruthenium(Ⅱ)complex for two-photon absorption-based optical power limiting in the near-IR range [J].Phys.Chem.Chem.Phys.,2011,13:17304.
[5] Gallavardin T,Maurin M,Marotte S,et al.Photodynamic therapy and two-photon bio-imaging applications of hydrophobic chromophores through amphiphilic polymer delivery[J].Photochem.Photobiol.Sci.,2011,10:1216.
[6] Hai Y,Chen J J,Zhao P,et al.Luminescent zinc salen complexes as single and two-photon fluorescence subcellular imaging probes[J].Chem.Commun.,2011,47:2435.
[7] Wang C K,Macak P,Luo Y,et al.Effects ofπ centers and symmetry on two-photon absorption cross sections of organic chromophores [J].J.Chem.Phys.,2001,114(22):9813.
[8] He G S,Tan L S,Zheng Q,et al.Multiphoton absorbing materials:molecular designs,characterizations,and applications [J].Chem.Rev.,2008,108:1245.
[9] Albota M,Beljonne D,Brédas J L,et al.Design of organic molecules with large two-photon absorption cross sections [J].Science,1998,281 (5383):1653.
[10] Li J,Gao Y,Huang X M,et al.Theoretical studies on nonlinear optical properties of triphenylamine derivatives[J].J.At.Mol.Phys.(原子与分子物理学报),2008,25(3):595(in Chinese)
[11] Tao L M,Zhao K,Sun Y H,et al.The solvent effect on two-photon absorption of N-{4-[(E)-2-(4-nitropheny1)viny1]phenyl}-N,N-diphenylamine[J].J.At.Mol.Phys.(原子与分 子物理学报),
2
004,21(1):111(in Chinese)
[12] Agou T,Hossain M D,Kawashima T,et al.Twophoton absorption properties of heterafluorenes and spirobiheterafluorenes bearing aminostyryl arms[J].Chem.Commun.,2009,(44):6762.
[13] Fukazawa A,Yamada H,Sasaki Y,et al.Zwitterionic ladder bis(arylethenyl)benzenes with large twophoton absorption cross sections[J].Chem.Asian J.,2010,5(3):466.
[14] Baumgartner T,Bergmans W,Kárpáti T,et al.From model compounds to extended π-conjugated systems:synthesis and properties of dithieno[3,2-b:2',3'-d]phospholes[J].Chem.Eur.J.,2005,11(16):4687.
[15] Romero-Nieto C,Kamada K,Cramb D T,et al.Synthesis and photophysical properties of donor-acceptor dithienophospholes [J].Eur.J.Org.Chem.,2010,27:5225.
[16] GAUSSIAN 09,References in http://www.gaussian.com/.
[17] DALTON,a molecular electronic structure program,Release Dalton2011(2011),see http://daltonprogram.org/.
[18] Li J,Sun Y P,Li Z L,et al.Ab initio study of one-photon and two-photon absorption properties of 2,5-bis[4-(2-arylvinyl)phenyl]-1,3,4-oxadiazoles[J].Chem.Phys.Lett.,2008,464:9.
猜你喜欢
现代电子技术(2020年7期)2020-06-15 06:42:00
火工品(2019年6期)2019-06-05 02:35:44
电子测试(2018年18期)2018-11-14 02:30:36
厦门大学学报(自然科学版)(2018年2期)2018-04-11 07:07:35
电力科学与工程(2016年5期)2016-07-04 09:17:06
环球飞行(2016年3期)2016-04-29 00:44:03
军民两用技术与产品(2016年3期)2016-03-26 08:06:00
当代化工研究(2016年5期)2016-03-20 16:21:32
中国医学装备(2015年10期)2015-12-29 12:00:22
原子与分子物理学报(2015年4期)2015-03-23 01:50:40
