
尿嘧啶-巴比妥酸分子间相互作用的密度泛函理论研究
2014-03-15 07:03:58裴玲
井冈山大学学报(自然科学版) 2014年1期
裴 玲
尿嘧啶-巴比妥酸分子间相互作用的密度泛函理论研究
裴 玲
(滨州学院化学与化工系,山东,滨州 256603)
采用密度泛函理论(DFT)中的B3LYP方法,在6-311G**理论水平上研究了尿嘧啶(U)和巴比妥酸(B)分子间的相互作用,以期从理论上预测巴比妥酸类物质对核酸产生的影响。优化复合物构型得到5个B1-U、4个B2-U、4个B3-U稳定构型,经基组重叠误差和零点振动能校正后,比较各复合物的相互作用能,可以得出复合物的主要存在形式有B2-U-3、B2-U-1、B1-U-3和B1-U-1。从NBO的分析得知B1-U、B2-U、B3-U系列复合物的稳定性顺序与氢键强度一致,即氢键的强度决定了复合物的稳定性,分析自然键轨道能揭示相互作用的本质。
尿嘧啶;巴比妥酸;密度泛函理论;相互作用能;自然键轨道
近年来金属离子、自由基、小分子化合物与碱基作用影响核酸结构及功能的实验和理论研究颇为活跃[1],尤其是药物分子与核酸基本单元相互作用的实验理论研究。巴比妥酸经常作为多种药物的前驱体,其衍生物在治疗多种疾病中有着广泛的应用[2]。赵剑英、张宇[3]用密度泛函理论研究了胞嘧啶一巴比妥酸分子间相互作用,从理论上证实了巴比妥酸类物质对核酸产生的影响。林雪飞、王芬等[4]采用B3LYP方法对胸腺嘧啶、巴比妥酸单体进行了优化计算,通过对单体的分析发现胸腺嘧啶和巴比妥酸能够通过某种相互作用力形成稳定的复合物,并判断出了这种弱相互作用力是氢键。虽然已经有关于碱基和巴比妥酸分子间相互作用的研究,尿嘧啶作为构成RNA的四种碱基之一,其与巴比妥酸分子间相互作用的研究未见报道,所以本研究在查阅相关文献的基础上对此进行研究。通过理论计算的方法找到尿嘧啶-巴比妥酸复合物间的相互作用力是如何影响其结构特性的,并且用自然键轨道分析去证实这种作用的影响,能够更深入地去了解这些复合体,为人类对生物体内的一些现象有全新的解释提供可参考的信息。
1 计算方法
巴比妥酸可能有10个互变异构体[5],Felice等人通过实验测定及理论计算,认为巴比妥酸的主要存在形式是B1、B2有少量存在,而B3仅有微量存在[5-7]。因此本研究选取B1、B2、B3为巴比妥酸的研究对象,其结构见图3.1。利用Gaussian03软件采用密度泛函理论在6-311G**基组下对四种单体及尿嘧啶-巴比妥酸复合物的结构进行优化,得到稳定结构,然后再对得到的稳定构型进行NBO分析、BSSE校正。所有的优化计算都在PC机上完成。
2 结果与讨论
2.1 几何构型分析
尿嘧啶、巴比妥酸单体及尿嘧啶-巴比妥酸复合物优化后的几何构型如图1所示,B1、B2、B3代表巴比妥酸三种常见的构型,U代表尿嘧啶。两者形成的氢键均为O…H键,且键长均在氢键键长范围内,相应的几何参数见表1、2、3。
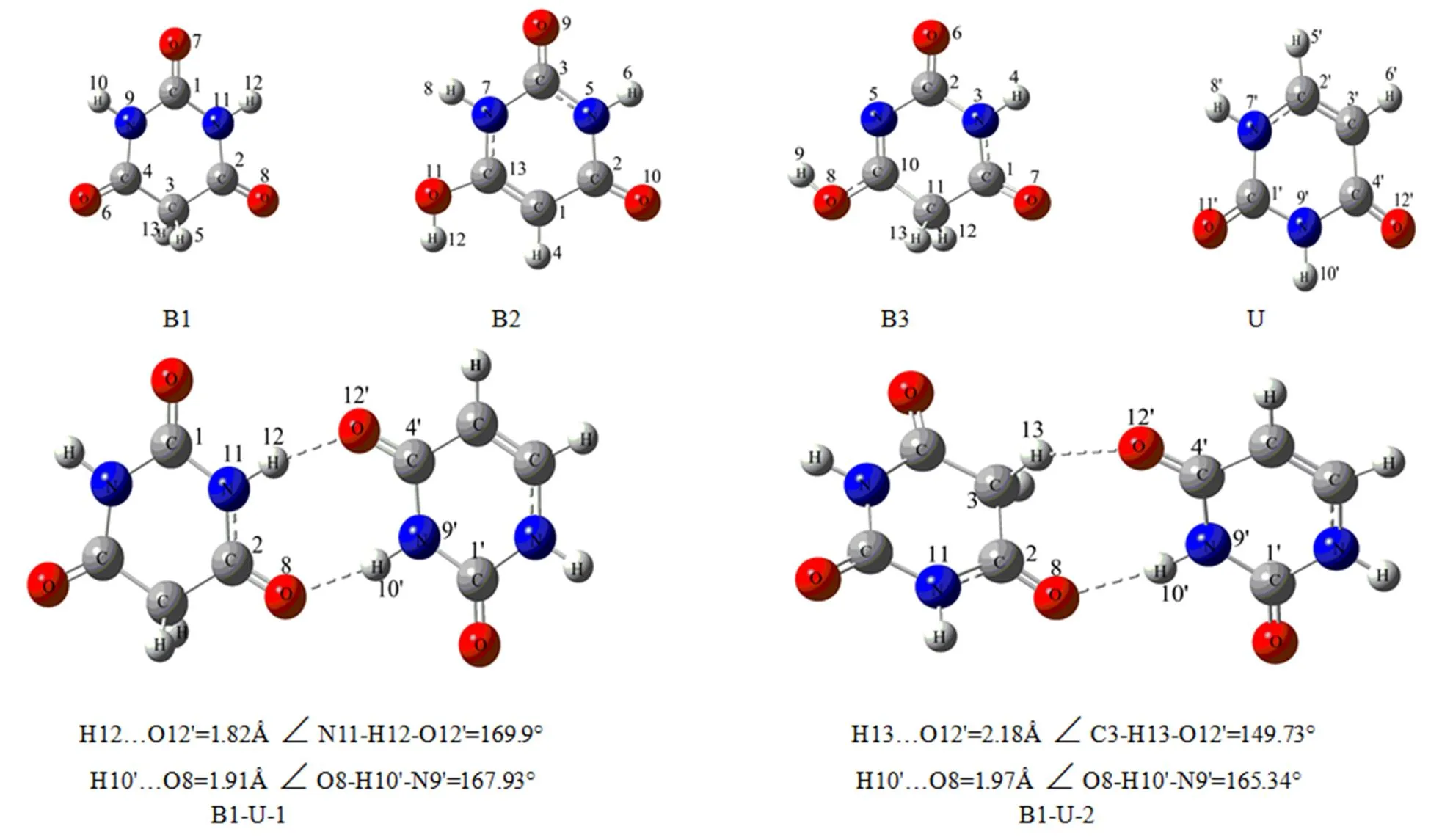
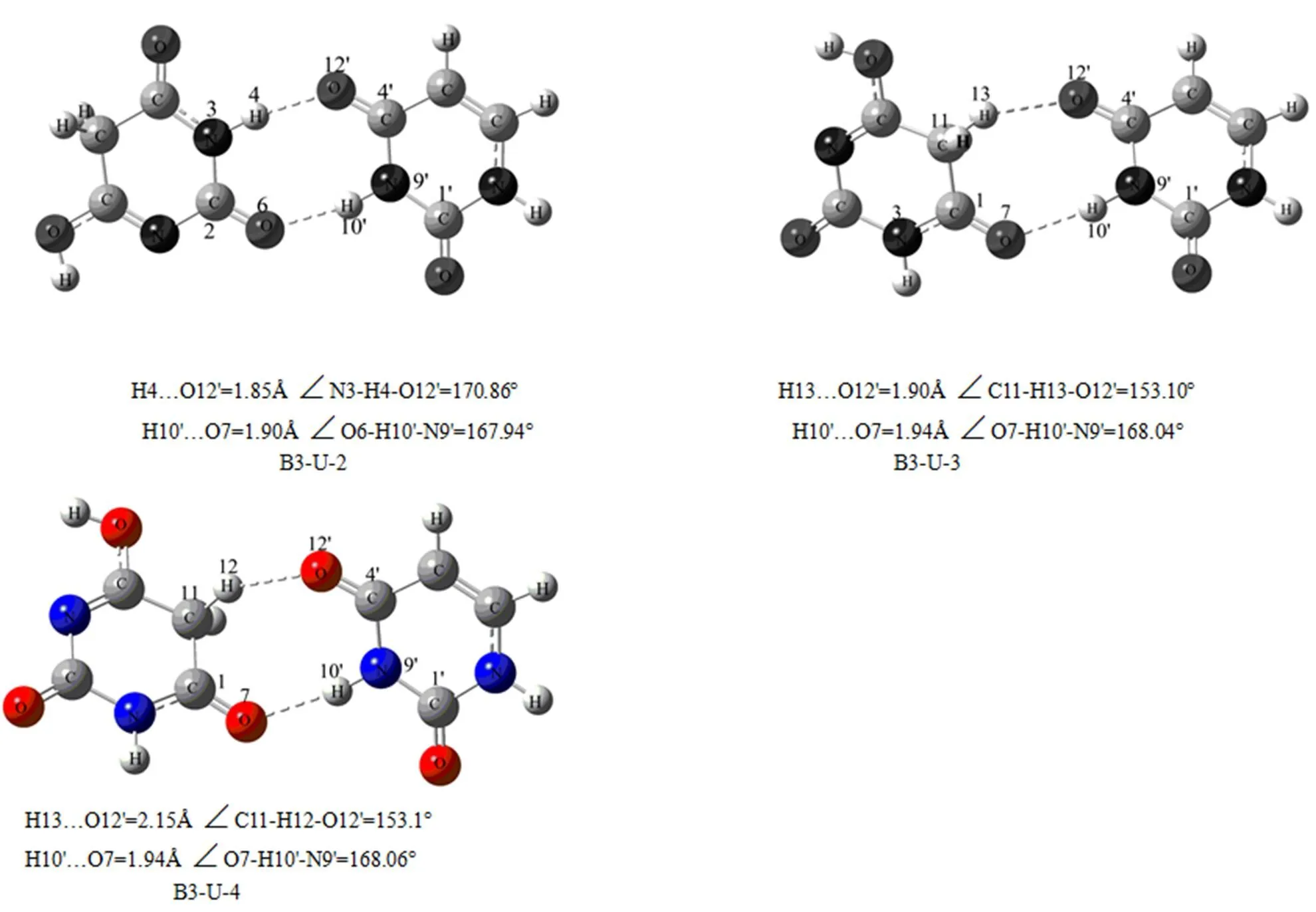
图1 尿嘧啶、巴比妥酸单体及复合物的优化构型图
2.1.1 B1-U复合物的几何构型
B1和U分子形成2个方向相反的氢键,这些氢键连接B1和U分子形成八元环,形成复合物前后,U环与B1键长均有变化,组成八元环的各键变化更明显,见表1。以B1-U-1构型为例,环上各键与单体相比,与氢键相邻者键增长,次邻者键缩短,即C3-H5、C4-O6、C4'-O12'、N9'-H10'键长分别增加了0.000052、0.000132、0.001379、0.001365 nm,而次邻者键C2-C3、C3-H13、C3-C4、C3'-C4'、N9-C4、N9'-C4'键长分别减少了0.000046、0.000159、0.000244、0.000663、0.000411、0.001577 nm。其余复合物中八元环上各键的变化趋势与此一致,比较该序列的键长变化。
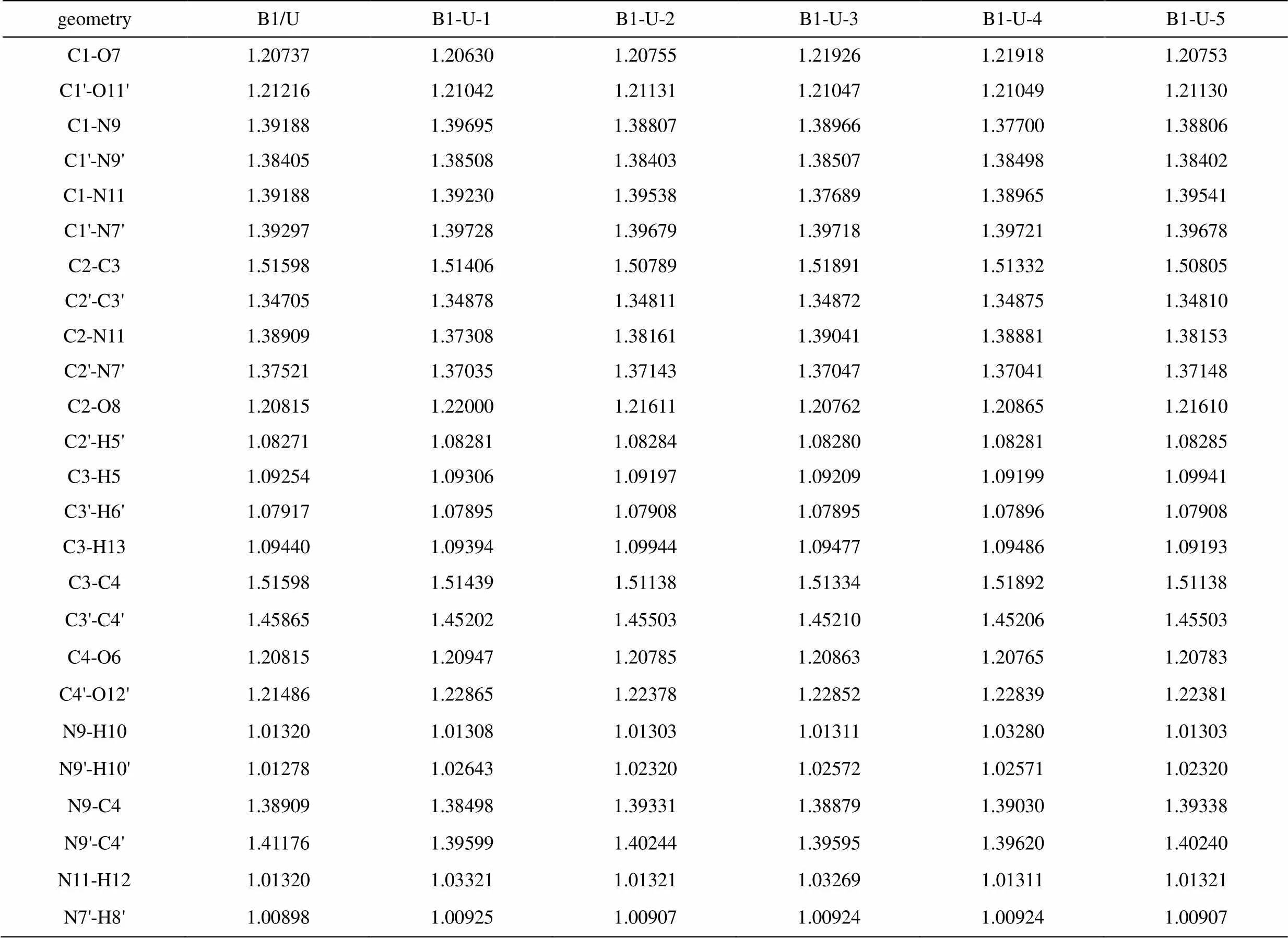
表1 尿嘧啶-巴比妥酸复合物(B1-U)的B3LYP/6-311G**部分优化几何参数(键长:Å)
O…H键长按B1-U-1、B1-U-4、B1-U-3、B1-U-2、B1-U-5的顺序增加,表明氢键的强度按此顺序减弱,比较八元环变化规律可以看出,形成的氢键强度越大,对相应八元环的影响越大。所以,可以得出复合物的稳定顺序为:B1-U-1 > B1-U-4 > B1-U-3 > B1-U-2 > B1-U-5。
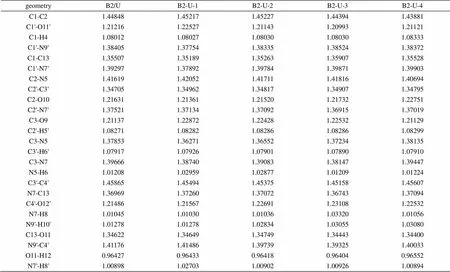
表2 尿嘧啶-巴比妥酸复合物(B2-U)的B3LYP/6-311G**部分优化几何参数(键长:Å)
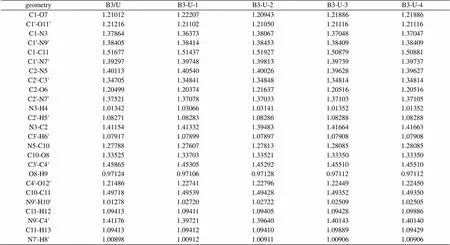
表3 尿嘧啶-巴比妥酸复合物(B3-U)的B3/LYP-6-311G**部分优化几何参数(键长:Å)
2.1.2 B2-U复合物的几何构型
与B1-U复合物相似,环上各键与单体相比,与氢键相邻者键增长,次邻者键缩短,虽说同系列的变化不是很有规律,但从大体趋势可以看出,在此复合物的几何构型中,氢键的形成对复合物中各键键长的影响情况与B1-U系列相似,见表2。O…H键长按B2-U-3、B2-U-1、B2-U-2、B2-U-4的顺序增加,所以复合物的稳定顺序为:B2-U-3 > B2-U-1 > B2-U-2 > B2-U-4。
2.1.3 B3-U复合物的几何构型
在此复合物的几何构型中,氢键的形成对复合物中各键键长的影响情况与B1-U系列相似,见表3。O…H键长按B3-U-2、B3-U-1、B3-U-3、B3-U-4,所以复合物的稳定顺序为:B3-U-2 > B3-U-1 > B3-U-3 > B2-U-4。
综合以上三个系列可以发现,虽然键长的变化幅度在每个构型中并没有很好的规律性,但大致的规律还是可以看出来的,所以我们可以得到各系列的稳定性顺序。另外,由平均氢键键长顺序可知三个序列中B2-U最稳定。
2.2 相互作用能
经过构型优化,得到各复合物分子的总能量(E)、零点能(ZPE)、基组叠加误差(BSSE)以及经ZPE和BSSE校正前后的分子间相互作用能(ΔE,∆EC,ZPE),见表4。将校正前和校正后的相互作用能相比较,可以看出,BSSE和ZPE对分子间相互作用能的重大影响,B2-U系列尤为明显,校正前后的分子间相互作用能相差很大。由此说明ZPE和BSSE的校正对于研究分子间的相互作用是非常必要的。由于所研究的对象主要是单体以氢键形成复合物分子,所以从结构上分析,几乎可将体系的相互作用能全部归诸于氢键的作用,并可由之推测氢键的平均键能。
对于B1-U复合物,B1-U-3的相互作用能最大,为41.266 kJ/mol,每个氢键的平均键能约为20.633 kJ/mol。B1-U-2的相互作用能最小,为26.254 kJ/mol,每个氢键的平均键能约为13.127 kJ/mol。由表3.4可以看出,ZEP和BSSE对B2-U系列的影响是最明显的,B2-U-3是这个序列里∆EC,ZPE最大的,为42.211 kJ/mol,氢键的平均键能约为21.1055 kJ/mol;B2-U-4最小,为28.203 kJ/mol,氢键的平均键能约为14.1015 kJ/mol。与B1-U系列相比较可知,B2-U较稳定,这与构型分析中的结论是一致的。对于B3-U系列的复合物,B3-U-2的∆EC,ZPE最大,为39.316 kJ/mol,氢键的平均键能约为19.658 kJ/mol;B3-U-3的∆EC,ZPE最小,为28.111 kJ/mol,氢键的平均键能约为14.0555 kJ/mol。依据总能量、相互作用能大小可知各复合物稳定性顺序为:B1-U-3≈B1-U-1≈B1-U-4 > B1-U-5≈B1-U-2,B2-U-3 > B2-U-1 > B2-U-2 > B2-U-4,B3-U-2 > B3-U-1 > B3-U-4≈B3-U-3,这与前面几何构型的分析结果大体是一致的,表明氢键强度对各类复合物的稳定性起关键性作用。
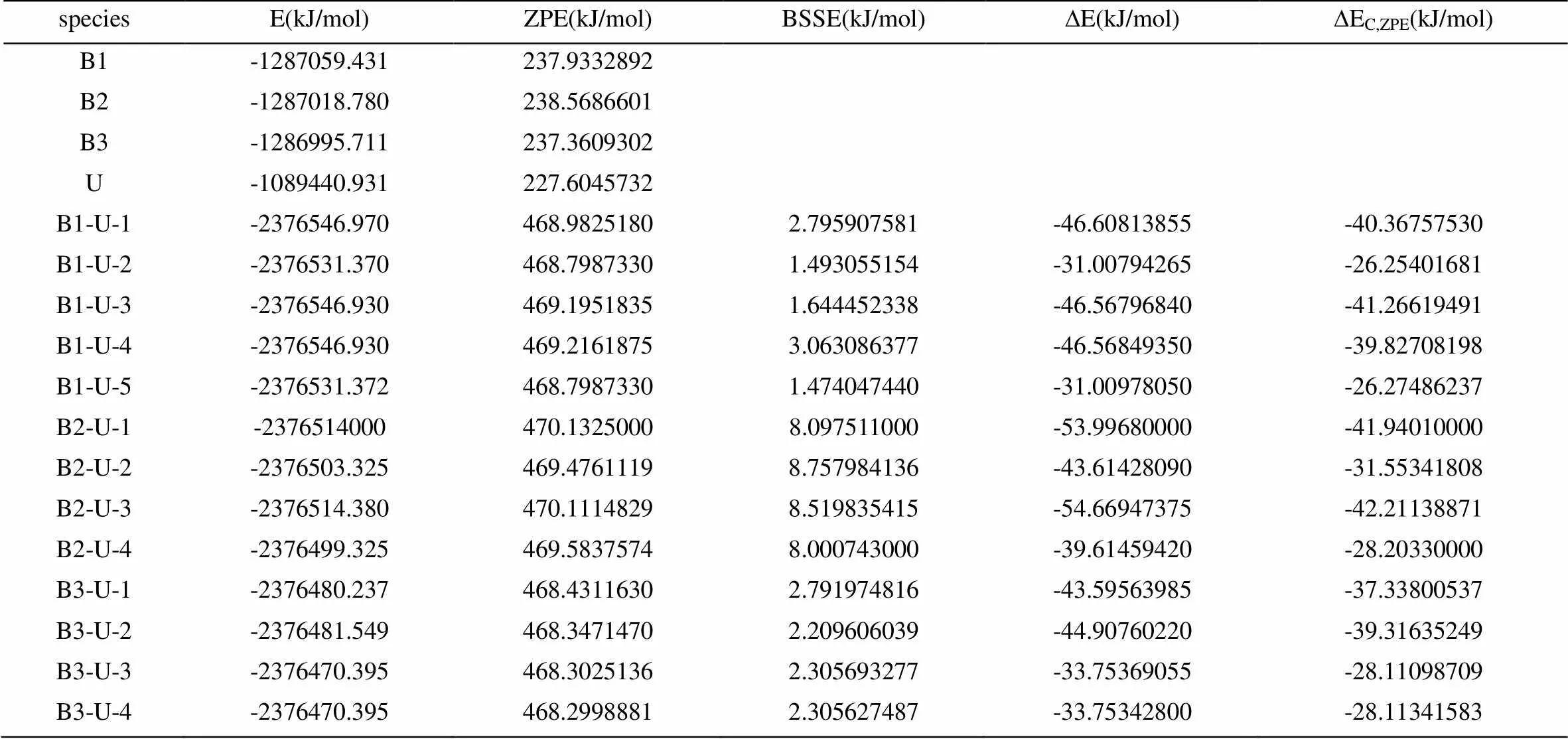
表4 B3LYP/6-311G**计算的总能量、零点能、基组叠加误差和相互作用能
2.3 自然键轨道分析
为了探讨分子间相互作用的本质,我们对B-U三个系列在B3LYP/6-311G**水平的自然键轨道(NBO)进行了分析。表5列出了电子供体(Donor)轨道i、电子受体(Acceptor)轨道j和它们之间相互作用的稳定化能E数值,E越大,表示i与j的相互作用越强,即i提供电子给j的倾向越大。
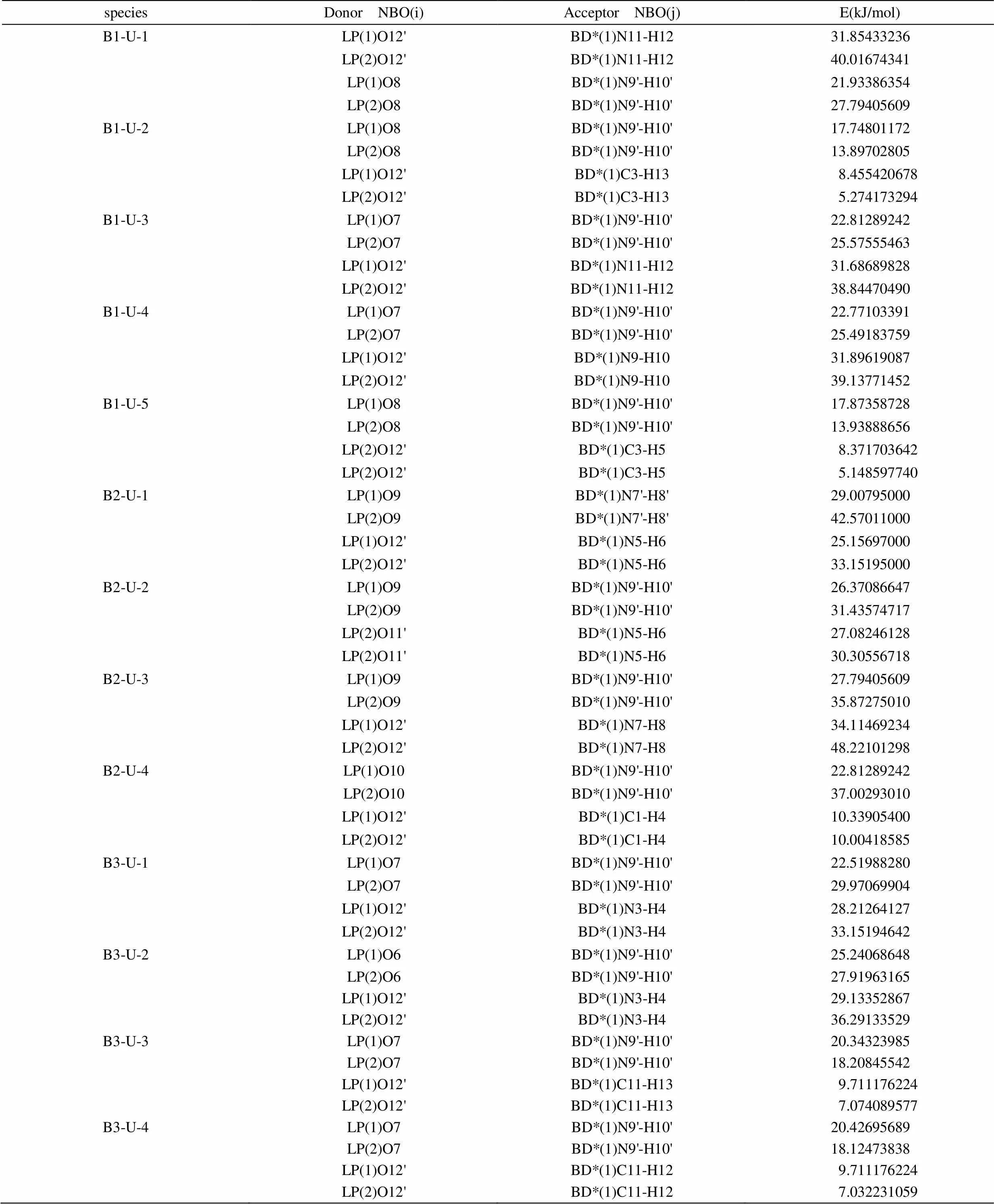
表5 尿嘧啶-巴比妥酸复合物的B3LYP/6-311G**自然键轨道分析
在B1-U系列中,最稳定构型B1-U-1中O12'的孤电子对(1)和(2)对N11-H12的反键轨道的稳定化能分别为31.854 kJ/mol和40.016 kJ/mol,O8的孤电子对(l)和(2)对N9'-H10'的反键轨道的稳定化能分别为21.933 kJ/mol和27.794 kJ/mol,这表明O12'-H12氢键较O8-H10'稳定,与图3.1中列出的氢键长短排序一致。该序列中,B1-U-1、B1-U-3、B1-U-4稳定性很相近,B1-U-2、B1-U-5稳定性很相近,且前一组的稳定性比后一组大得多。即稳定性大小排序:B1-U-1 > B1-U-4 > B1-U-3 > B1-U-2 > B1-U-5,这与几何构型分析的结果基本一致。
对于B2-U复合物,各复合物中相应氢键的稳定化能大小与该键长次序一致,而且此序列的平均氢键稳定化能大于B1-U系列的,说明B2-U系列的复合物比B1-U系列的稳定,比较数据可得出B2-U系列复合物稳定性大小排序:B2-U-3﹥B2-U-1 > B2-U-2 > B2-U-4,这也与几何构型分析的结果是一致的。
对于B3-U复合物,各复合物中相应氢键的稳定化能大小与该键长次序一致,各构型总稳定化能之和的排序与几何构型分析的结果也是一致的。
以上对复合物做的NBO分析表明,复合物稳定性由氢键强度决定。
3 结论
在B3LYP/6-311G**水平上计算尿嘧啶和巴比妥酸单体和复合物,发现分子间的相互作用基本没有改变尿嘧啶和巴比妥酸的分子构型,只是各单体的键长有微小的变化,两分子间主要以氢键结合,均形成了八元环。从对相互作用能和NBO的分析来看,B1-U、B2-U、B3-U系列复合物的稳定性顺序与氢键强度一致,也就是说,氢键的强度决定了复合物的稳定性。对于B1-U系列存在的不同构型稳定性相近的情况,主要是因为他们几何构型的相似性。比较各复合物的相互作用能,可以得出复合物的主要存在形式有B2-U-3、B2-U-1、B1-U-3和B1-U-1。
[1] 林雪飞, 孙成科, 杨思娅, 等. 6-烷基鸟嘌呤与DNA碱基间氢键作用的理论研究[J]. 化学学报, 2010, 68(5):326-345.
[2] Robert A A. Thermodynamic properties of enzyme-catalyzed reactions involving cytosine, uracil, thymine, and their nucleosides and nucleotides[J]. BiophyChem, 2007, 127(1-2): 91-96.
[3] 赵剑英, 张宇. 胞嘧啶-巴比妥酸分子间相互作用的密度泛函理论研究[J]. 计算机与应用化学, 2009, 26(2):209-216.
[4] 林雪飞, 王芬, 张水花, 等. 胸腺嘧啶-巴比妥酸分子间相互作用的密度泛函理论研究[J]. 曲靖师范学院学报, 2011, 30(6):36-44.
[5] Zuccarello F, Buemi G , Gandolf C, et al. Barbituric and thiobarbituric acids:a conformational and spectroscopic study[J]. Spectrochm Acta Part A, 2003, 59(1):139-151.
[6] Racnondo F, Pieretti A, Gontrani L. Hydrogen bonding in barbituric and 2-thiobarbituric acids: a theoretical and FT-IR study[J]. Chen. Phy, 2001, 271(3):293-308.
[7] Ralhan S, Ray N K. Density functional study of barbituric acid and its tautomers[J]. J Mol Struc (Theochem), 2003, 634(1-3):83-88.
DENSITY FUNCTIONAL THEORY ON THE INTERACTION OF URACIL AND BARBITURIC ACID
PEI Ling
(Department of Chemistry and Chemical Engineering, Binzhou University, Binzhou, Shandong 256603, China)
In this paper the interaction between uracil(U) and barbituric acid(B) was studied by using the density functional theory (DFT) at the B3LYP/6-311G** level, for the purpose of further investigating the effect of barbituric acid on the structure of nucleic acid. The geometries were optimized and five B1-U, four B2-U and four B3-U stable complexes were obtained. Comparing the interaction energies after the correction of the basis set superposition error and zero point energies, it may show that the main form of the compound were B2-U-3, B2-U-1, B1-U-3 and B1-U-1. The stability of the series of complexes were consistent with the hydrogen bonding strength from the NBO analysis, namely hydrogen bonding strength determined the stability of the complexes, and natural bond orbital analysis could reveal the nature of interaction.
uracil; barbituric acid; density functional theory; interaction energies; natural bond orbital
1674-8085(2014)01-0021-08
O641
A
10.3969/j.issn.1674-8085.2014.01.005
2013-10-10;
2013-12-22
裴 玲(1980-),女,山东邹平人,讲师,硕士,主要从事电化学和量化理论计算研究(E-mail: peiling1201@163.com).
猜你喜欢
中学生理科应试(2024年1期)2024-05-18 13:02:52
核农学报(2020年2期)2020-03-11 08:33:16
青岛大学学报(工程技术版)(2019年2期)2019-09-10 07:22:44
菏泽学院学报(2017年2期)2017-05-16 08:59:11
陕西师范大学学报(自然科学版)(2015年1期)2016-01-16 03:23:30
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
中学化学(2015年8期)2015-12-29 07:32:44
哈尔滨商业大学学报(自然科学版)(2014年5期)2014-09-14 04:32:28
原子与分子物理学报(2014年3期)2014-02-28 22:18:23
