
Cr+3对乙醛C—C键激发脱羰基反应的量化计算研究
2014-03-14 01:08:44刘红飞
精细石油化工 2014年5期
刘红飞
(宁夏大学物理电气信息学院,宁夏 银川 750021)
脱羰基反应是石油化工生产中的重要的反应之一, 反应中C—C和C—H键的激发是研究的重点[1-3]。人们已经对Cr+、Fe+和Co+对小分子醛的脱碳反应作了大量的实验研究[4-9]。Sonnenfroh等[10]通过实验提出了反应机理(见图1), 但是没有确定该反应的具体路径是通过C—C激发还是由C—H激发的,同时所有基元反应势能面的具体信息也不清楚。为此,笔者以乙醛为模型,Cr+3作为催化剂,利用密度泛函理论,对脱羰基反应机理进行了理论研究,明确反应的激发反应机理,并确定其反应的中间体、过渡态、能量来确定其具体的反应路径。
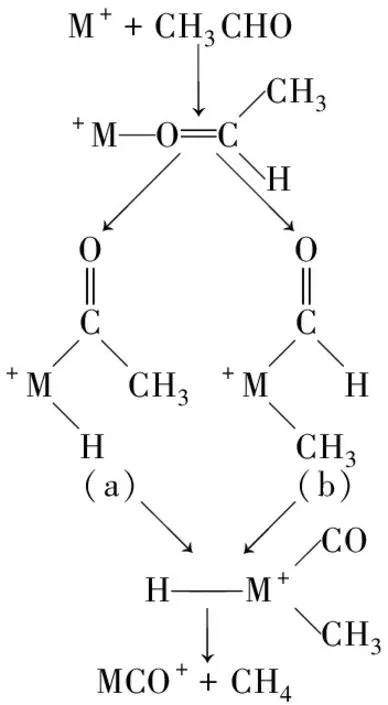
图1 早期研究金属离子对CH3CHO脱羰基反应的机理
1 计算方法
采用基于DFT的B3LYP方法,计算全部使用Gaussion03程序[11]。Cr原子使用Lanl2dz基组[12-13],C、H、O使用6-31+G*基组,此法已经被广泛应用于过渡金属及其化合物与有机分子反应的开壳层体系的电子结构计算,并被证明为既能满足够的精度又具有合理的计算效率。计算振动频率,得到零点能修正(ZPE),用来判定所优化的结构是中间体还是过渡态(中间无虚频,过渡态只有1个虚频)。过渡态与所连接的两个极小值的关系由过渡态的虚频矢量的方向判断,通过内禀反应坐标(IRC)计算确认反应中间体和反应路径[14]。
2 结果及讨论
根据计算结果,通过对中间体和过渡态的结构和能量分析发现,整个反应经历四个阶段:开始结合、C—C激发、H转移和解离。CH3CHO要经过两个中间体和两个过渡态才能完成脱羰反应,首先CH3CHO和Cr+3初始结合,Cr1和CH3CHO通过和O2吸附结合,此时,Cr1的净电荷为2.057,Cr1和O2的距离为0.189 7 nm,C3和O2之间的距离为0.125 9 nm,能量比反应物能量下降了167.7 kJ/mol。
第一个过渡态TS1,是由O2的电荷向Cr1转移,从-0.323变为-0.101,C3的电荷基本保持不变所形成的,这时ΔE1为167.7 kJ/mol,金属离子Cr+3插入乙醛的C3—C5键中。具有Cs对称结构的,其对称面为O2—C3—Cr1—C5, Cr1—H6、Cr1—C5、Cr1—C3和Cr1—C2的键长分别为0.236 2,0.205 4,0.213 1 nm和0.191 3 nm,Cr+3的s和d轨道具有较好的sd轨道杂化。表明中心原子Cr1与这些原子具有较强的作用,过渡态TS1是基态势能面上连接着中间体M1和M2的一阶鞍点,和中间体M2所对应的键长十分接近,是势能面上一个晚期过渡态。过渡态的能量比初始反应物低76.9 kJ/mol,比与它相连的中间体M1和中间体M2高244.6 kJ/mol和18.8 kJ/mol。

表1 Cr+3 对CH3CHO脱羰反应各鞍点的相对能

图2 Cr+3对CH3CHO脱羰反应各驻点的分子几何构型
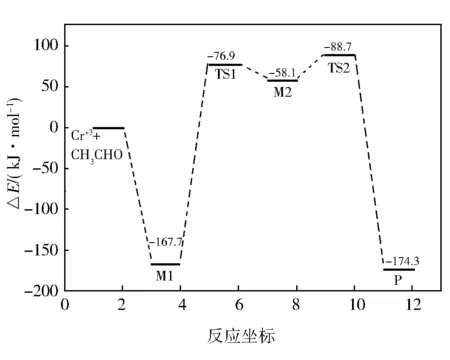
图3 Cr+3 对CH3CHO脱羰反应过程各驻点能级变化示意
第二个中间体M2,能量较TS1降低18.8 kJ/mol,从分子结构图中可以看出,金属离子Cr+3继续插入乙醛的C3—C5键中。M2具有Cs对称性,其对称面为O2—C3—Cr1—C5,Cr+3的s和d轨道具有较好的sd杂化。Cr1—H6、Cr1—C5、Cr1—C3和 Cr1—C2的键长分别为0.229 6,0.197 2,0.201 2 nm 和0.195 1 nm,表明中心原子Cr1与这些原子仍然具有较强的作用,C3—C5的 距离为0.318 7 nm,意味着C3—C5键已经断裂,生成Cr+3CH3和CHO,并经过TS2的反应生成最终产物CH4—Cr+3—CO。
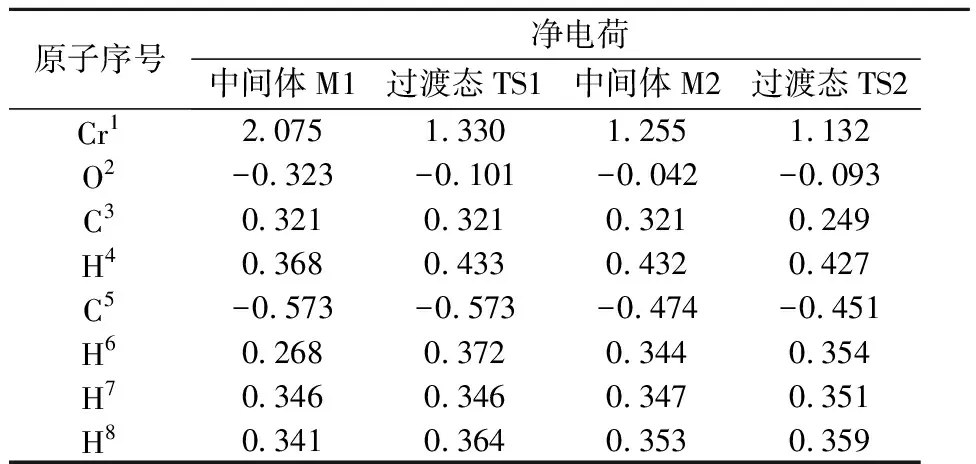
表2 中间体和过渡态的净电荷分布
中间体M2和产物P通过H4转移到鞍点TS2进行连接。从结构上看,C3—C5和C3—H4键长均变长,C5—H4键长缩短,Cr1的继续获得电荷为1.132,O2则开始获得电荷从-0.042增加到-0.093,C3则继续失去电荷从0.321变为0.249,H4—C3之间的距离增加到0.121 2 nm,H4离开O2,向C5靠近,C5失去电荷和H4作用增强。同时说明过渡态连按的一侧是最终产物P。最后由M2经历TS2后生成产物,CH4和CO,和第二个过渡态的ΔE2为88.7 kJ/mol,在这两个ΔE中,第二步反应的ΔE最大,是反应中最重要的一步。
Sonnenfroh[10]和Khan[15]分别用离子束在试验中发现了Cr+3—CO和计算结果吻合,表明所选择的计算方法对于反应出口相对能量的计算还是比较可靠的。因此,所选的理论方法能准确描述势能面的特征。同时,反应经历的四个阶段中,最大的活化能是88.7 kJ/mol,比较低,反应很容易实现,表明Cr+3具有较好的催化效果。
3 结 论
a.脱羰基过程主要是由C—C键激发所产生的,而不是C—H键激发,整个反应经历四个阶段:开始吸附结合、C—C键激发、H原子转移和分子解离。
b.势能面的分析表明CH3CHO要经过两个中间体和两个过渡态才能完成反应,第一个过渡态TS1的ΔE1为76.9 kJ/mol,第二个过渡态TS1的ΔE2为88.7 kJ/mol,生成产物CH4和CO,第二个过渡态的反应是最主要的环节。
参 考 文 献
[1] Cassady C J, Freiser B S. Gas-phase reactions of transition-metal ions with methyl nitrite and nitromethane[J]. J Am Chem Soc, 1985,107(6):1566-1573.
[2] Liu Chengbu, Zhang Dongju, Bian Wensheng. Theoretical investigation of the reactions of Co+with OCS[J]. J Phys Chem A, 2003,107:8618-8622.
[3] Zhao Lianming, Zhang Rongrong, Guo Wenyue, et al. Does the Co+-assisted decarbonylation of acetaldehyde occur via C—C or C—H activation: A theoretical investigation using density functional theory[J]. Chem Phys Lett, 2005,414(1-3): 28-33.
[4] 邵国泉,方维海,陈光巨,等.丙烯醛及其衍生物基态脱羰基反应机理的理论研究[J],物理化学学报,1996,12(9):830-835.
[5] 林玲,丁万见,方维海,等.硫代羰基化合物激发态结构及光化学反应的理论预示[J],化学学报,2003,61(1):1-7.
[6] Corderman R R, Beauchamp J L. Ion cyclotron reaonance investigation of the decarbonylation of aldehydes by (.eta.5-cyclopentadienyl)nickel(1+)[J]. J Am Chem Soc, 1976,98(18):5700-5702.
[7] Halle L F, Crowe W E, Armentrout P B. et al. Reactions of atomic cobalt ions with aldehydes and ketones: Observation of decarbonylation processes leading to formation of metal alkyls and metallacycles in the gas phase[J]. Organometallics, 1984,3(11):1694-1706.
[8] Tolbert M A, Beauchamp J L. Homolytic and heterolytic bond dissociation energies of the second row group 8,9,and 10 diatomic transition-metal hydrides: Correlation with electronic structure[J]. J Phys Chem,1986,90(21):5015-5022.
[9] Liu Haichuan, Hu Yihua, Yang Shihe, et al. Experimental and computational studies of intra-complex reactions in Mg+(primary, secondary alkylamine) induced by photoexcitation of Mg+[J]. J Chem Eur, 2005,11(21): 6392-6406.
[10] Sonnenfroh D M, Farrar J M.Crossed-beam studies of energy and allgular distributions of organometallic reaction decarbonylation of acetaldehyde by Fe+and Cr+[J].J Am Chem Soc,1986,108:3521-3522.
[11] Frisch M J, Trucks G W, Schlegel H.B, et al. Gaussian 03 (revision B.05). Pittsburgh, PA: Gaussian, Inc, 2003.
[12] Zhang Dongju, Liu Chengbu, Bian Wensheng. Theoretical investigation of the reactions of Fe+with OCS[J]. J Phys Chem A, 2003,107:8955-8960.
[13] Carpenter C J, Koppen M P A van, Bowers M T. Details of potential energy surfaces involving C—C Bond activation: Reactions of Fe, Co, and Ni with acetone[J]. J Am Chem Soc,1995,117:10976-10985.
[14] Hay P J, Wadt W R. Ab inito effective core potentials for molecular calcuations: potentials for the transition metal atoms Sc to Hg[J]. J Chem Phys,1985, 82(1):270-283.
[15] Khan F A, Clemmer D E, Schulta R H, et al. Sequential bond energies of chromiums (Cr(CO)x+,x=1~6)[J]. Phys Chem,1993,97:7978-7987.
猜你喜欢
北京航空航天大学学报(2022年5期)2022-06-06 09:27:18
陶瓷学报(2021年5期)2021-11-22 06:35:34
大学化学(2021年8期)2021-09-26 10:51:16
青岛大学学报(工程技术版)(2019年2期)2019-09-10 07:22:44
电脑知识与技术(2018年3期)2018-03-21 09:27:04
哈尔滨理工大学学报(2017年1期)2017-04-08 04:16:24
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
中学化学(2015年8期)2015-12-29 07:32:44
应用化工(2014年1期)2014-08-16 13:34:08
