
Molecular detection of Xanthomonas oryzae pv.oryzae,Xanthomonas oryzae pv.oryzicola,and Burkholderia glumae in infected rice seeds and leaves
2014-03-13 05:51:10WenLuLuqiPnHijunZhoYulinJiYnliWngXiopingYuXueynWng
The Crop Journal 2014年6期
Wen Lu,Luqi Pn,Hijun Zho,Yulin Ji,Ynli Wng ,Xioping Yu,Xueyn Wng,*
aZhejiang Provincial Key Laboratory of Biometrology and Inspection & Quarantine,College of Life Science,China Jiliang University,Hangzhou 310018,China
bInstitute of Nuclear-Agricultural Science,Zhejiang University,Hangzhou 310029,China
cUnited States Department of Agriculture,Agricultural Research Service,Dale Bumpers National Rice Research Center(USDA-ARS DB NRRC),Stuttgart,AR,USA
dInstitute of Plant Protection and Microbe,Zhejiang Academy of Agricultural Sciences,Hangzhou 310021,China
1.Introduction
Rice,one of the most important food crops,is constantly challenged by bacterial pathogens,such as those causing bacterial blight,leaf streak,and bacterial panicle blight.Bacterial blight,caused by Xanthomonas oryzae pv.oryzae,is a prevalent and destructive rice disease that causes annual yield losses ranging from 10 to 20% and up to 50% to 70% in severely infected fields [1,2].This disease also affects grain quality by interfering with the maturation process[3].Bacterial leaf streak caused by X.oryzae pv.oryzicola,the pathovar of X.oryzae pv.oryzae,usually results in the wilting of leaves andlosses as high as 32%in 1000-grain weight[4].It is important to note that hybrid rice varieties are more susceptible to this bacterial pathogen than non-hybrid varieties [5].Rice bacterial panicle blight(bacterial grain rot),caused by Burkholderia glumae was first reported in Japan in 1956 [6].Yield losses due to B.glumae can reach as high as 40%in the southern U.S.[7].Given that the optimal temperature for the growth of B.glumae ranges from 30 to 50 °C [7],warmer temperatures during the ricegrowing season increase the severity of the disease [8].The presence of X.oryzae pv.oryzae,X.oryzae pv.oryzicola,B.glumae in infected seeds may cause disease transmission,so that many countries have listed the three bacteria as quarantined
organisms.Both conventional and real-time PCR have been widely used to detect or verify the presence of X.oryzae pv.

Table 1-Sequences,annealing temperature,predicted product size,primers,and primer sources used in this study.

Table 2-Bacterial and fungal strains used for specificity tests.
oryzae[9–13],X.oryzae pv.oryzicola[14–16],and B.glumae[17–20]in recent decades.These molecular-based methods are rapid,accurate and sensitive for detecting pathogens.However,they can detect only one pathogen each.Several methods have been developed to distinguish highly similar pathovars of X.oryzae pv.oryzae and X.oryzae pv.oryzicola using multiplex or real-time PCR[21,22].
In the present study,we used genome sequence information available in public databases to develop PCR primers for accurate identification of X.oryzae pv.oryzae,X.oryzae pv.oryzicola,and B.glumae.The objective of this study was to develop multiplex PCR and SYBR Green real-time PCR methods for simultaneous detection of the presence of X.oryzae pv.oryzae,X.oryzae pv.oryzicola,and B.glumae.
2.Materials and methods
2.1.Bacterial and fungal strains and culture conditions
Strains of X.oryzae pv.oryzae,X.oryzae pv.oryzicola,and B.glumae; the other closely related pathogens Xanthomonas campestris,Xanthomonas maltophilia,Burkholderia gladioli pv.alliicola,and Burkholderia cepacia;and the rice fungal pathogens Magnaporthe oryzae and Ustilaginoidea oryzae were used to develop specific primer sets.Bacterial strains were cultured on a Luria–Bertani medium (1% tryptone,0.5% yeast extract,1%NaCl,and 1.5%agar)at 28 °C for two days.Fungal isolates were cultured on corn meal medium(3%corn meal and 1.5%agar)at room temperature for four to five days[23].
2.2.DNA preparation
Genomic DNA of bacterial strains was extracted with a Genomic DNA Prep Kit (Sangon,Shanghai,China) following the manufacturer's protocol,except that DNA was eluted in 30 μL double-distilled water(ddH2O).Genomic DNA of fungal and leaf tissue was prepared using the CTAB method [24,25].DNA concentrations were measured with a Nanodrop 2000 instrument (Thermo Fisher Scientific,Wilmington,DE).The OD260:OD280 ratios of all samples were approximately 1.8.All samples were diluted to 1 ng μL-1in ddH2O.
2.3.Development of specific DNA primers
The sequence of the putative glycosyltransferase gene of X.oryzae pv.oryzae (AF169030.1) was identified in GenBank,and then aligned with the putative glycosyltransferase genes of X.oryzae pv.oryzicola (CP003057.1),X.campestris pv.campestris (AF204145.1),X.campestris pv.vesicatoria(AM039952.1),Xanthomonas axonopodis pv.citrumelo(CP002914.1),and Xanthomonas albilineans(FP565176)using BioEdit[26].Specific primers for X.oryzae pv.oryzae were designed from non-conserved regions (Table 2,Fig.S1).Using the same strategy,the AvrRxo gene of X.oryzae pv.oryzicola(AY395713.1) was used as a template for designing specific primers for X.oryzae pv.oryzicola (Fig.S2).Ribosomal internal transcribed spacers (ITSs) of B.glumae (D87080),B.plantarii(AB183680.1),B.gladioli (EF552066.1),B.gladioli pv.alliicola(D87082.1),B.gladioli pv.agricicola (EF552068.1),and B.cepalia(FJ870551.2)were aligned,after which the nonconserved regions were used to design specific primers(Fig.S3).The PCR product lengths ranged from 100 to 250 bp for both conventional and real-time PCR assays.
2.4.Polymerase chain reaction (PCR)
Conventional PCR assays were used to test the specificity and sensitivity of primers using a T100 Thermal Cycler (Bio-Rad,California,USA).The concentration of the sample used for testing the specificity of the primers was 1 ng μL-1.The pathogens X.oryzae pv.oryzae OS198,X.oryzae pv.oryzicola AHB4-75,and B.glumae LMG2196 were diluted to 5 × 10-1,1 × 10-1,5 × 10-2,1 × 10-2,5 × 10-3,1 × 10-3,5 × 10-4,and 1 × 10-4ng μL-1with ddH2O to test primer sensitivity.PCR reactions were performed in a final volume of 20 μL containing 10 μL of 2 × Taq master mix(Sangon,Shanghai,China),0.4 μL of each 10 μmol L-1primer,1 μL of genomic DNA,and 8.6 μL ddH2O,vortexed thoroughly.PCR amplification was as follows:initial denaturation for 3 min at 94 °C;35 cycles of 30 s at 94 °C,30 s at 58 °C,30 s at 72 °C,and final extension for 10 min at 72 °C.PCR products were separated on a 1%agarose gel(1 × TAE buffer)by electrophoresis at 100 V for 30 min and visualized with a Gene Genius Bio Imaging System (Syngene,Cambridge,UK).DNA templates were replaced with ddH2O as a negative control.
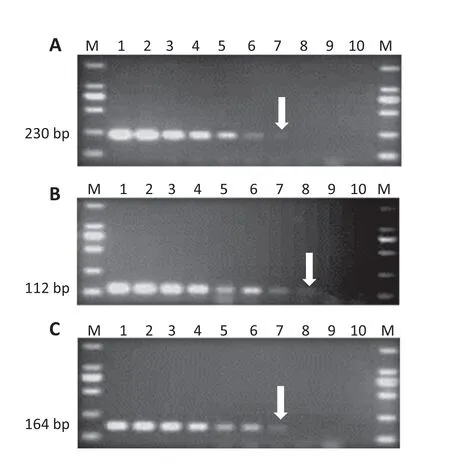
Fig.1-Sensitivity tests of primer sets using conventional PCR.A:sensitivity test of JLXooF/R with the template OS198;B:sensitivity test of JLXocF/R with the template AHB4-75;C:sensitivity test of JLBgF/R with the template LMG2196.Lane M,DNA ladder(DL 2000,Takara,Shiga,Japan);lanes 1-9:1,5 × 10-1,1 × 10-1,5 × 10-2,1 × 10-2,5 × 10-3,1 × 10-3,5 × 10-4,and 1 × 10-4 ng μL-1;lane 10:negative control.The arrows point to the limiting detection concentrations of the primer sets.
2.5.SYBR Green real-time PCR
The SYBR Green real-time PCR assay was used to test the sensitivity of the primers with an IQ5 Multicolor real-time PCR Detection System (Bio-Rad,Hercules,CA).DNA of OS198,AHB4-75,and LMG2196 was 10-fold serially diluted from 1 to 1 × 10-6ng μL-1.Each PCR reaction contained 10 μL of 2 × SYBR Premix Ex Taq (TaKaRa,Shiga,Japan) and 0.4 μL of each 10 μmol L-1primer,1 μL template,and 8.6 μL ddH2O.Realtime PCR was performed with the following program: 45 s at 95 °C;40 cycles of 5 s at 95 °C,30 s at 61 °C for 30 s;and melting curve at 65 to 95 °C with increases of 0.5 °C.DNA templates were replaced by ddH2O as a negative control.
2.6.Multiplex PCR
To perform multiplex PCR,1 ng μL-1genomic DNA of OS198,AHB4-75 and LMG2196 was used as positive templates in three PCR tubes,respectively.The three genomes were mixed with different concentrations and proportions of DNA to test the primers' sensitivity in a multiplex PCR reaction.The total volume of multiplex PCR was 20 μL (10 μL of 2 × Taq master mix,0.4 μL of 10 μmol L-1of each primer,and 1 μL DNA mix).PCR products were separated on a 1.5%agarose gel (1 × TAE buffer) by electrophoresis at 90 V for 50 min and visualized with the Gene Genius Bio Imaging System.DNA templates were replaced by ddH2O as a negative control.

Fig.2-Sensitivity tests of JLXooF/R primer set using SYBR Green RT-PCR.A: Standard curve.For each assay,templates (1-7)were diluted 10-fold to concentrations ranging from 1.0 to 1.0 × 10-6 ng μL-1.B: Melting-peak analysis.C: Fluorescence intensity;1.0 to 1.0 × 10-6 ng μL-1;1-7:samples;8:negative control.The arrow points to the limiting detection concentration of the primer set;D:CT(cycle threshold)and SE (standard error).
2.7.Artificial inoculation of seeds with X.oryzae pv.oryzae,X.oryzae pv.oryzicola,and B.glumae
Five grams(approximately 150 seeds)of rice cultivar Nipponbare were surface-disinfected in 75%ethanol for 10 min,incubated in approximately 0.5% chlorine solution for 30 min,and rinsed three times with sterilized distilled water.After disinfection,the seeds were transferred to Petri dishes containing sterilized filter paper and allowed to air-dry for 3 h in a laminar-flow chamber.The surface-disinfected seeds were inoculated with 5 mL g-1of bacterial suspensions of OS198 or AHB4-75 or LMG2196 or a mixture of OS198,AHB4-75,and LMG2196 with OD600equal to 0.01(×108CFU mL-1),respectively.OD600values were measured using a Nanodrop (ND 100 spectrophotometer,NanoDrop Technologies,Inc.).The inoculation was vacuum infiltrated for 60 min.After inoculation,the artificially infected seeds were allowed to air-dry in the laminar air flow chamber and stored at 4until use.
2.8.The detection of pathogens on rice seeds
Detection of X.oryzae pv.oryzae,X.oryzae pv.oryzicola,and B.glumae in rice seed lots was performed by washing 1 g healthy and 1 g infected seeds infected by X.oryzae pv.oryzae,X.oryzae pv.oryzicola,B.glumae,or a mixture of the three bacteria in 5 mL sterile dH2O,shaking at 100 r min-1for 2 h at 4 °C.One microliter of suspension was used as the template for the multiplex PCR described above for detection of X.oryzae pv.oryzae,X.oryzae pv.oryzicola,and B.glumae.All experiments were repeated twice.
3.Results
3.1.Primer design and specificity
The specific primers JLXooF/R for X.oryzae pv.oryzae,JLXocF/R for X.oryzae pv.oryzicola,and JLBgF/R for B.glumae were developed based on the polymorphic regions of the corresponding putative glycosyltransferase gene,AvrRxo gene and ITS sequence,respectively(Table 1,Figs.S1,S2,and S3).The 230 bp DNA fragments were amplified from all X.oryzae pv.oryzae strains using the JLXooF/R.However,the expected fragments were not amplified either from closely related bacterial strains,including X.oryzae pv.oryzicola and X.campestris,or from other bacterial or fungal strains(Table 2,Fig.S4).An expected 112 bp DNA product was amplified only from X.oryzae pv.oryzicola strains using the primer set JLXocF/R (Table 2,Fig.S5),and a product of 164 bp was amplified only from B.glumae using JLBgF/R (Table 2,Fig.S6).The results suggest that these primer sets were specific to the target pathogens tested.
3.2.Sensitivity of PCR amplification
The purified DNA was used to test the primers' sensitivity in both conventional PCR and real-time PCR assays.The primer sets JLXooF/R,JLXocF/R,and JLBgF/R detected as little as 1 pg μL-1DNA of OS198,0.5 pg μL-1DNA of AHB4-75,and 1 pg μL-1DNA of LMG2196 in the 20 μL PCR reactions(Fig.1).
SYBR Green real-time PCR was also used to test the sensitivity of the primer sets.The amplification profiles of OS198,AHB4-75,and LMG2196 dilutions are shown in Figs.2,3,and 4,respectively.The R2values of JLXooF/R,JLXocF/R,and JLBgF/R were equal to 0.998,0.996,and 0.992,respectively,indicating a good linear response of each primer set.The linear regression slope gave coefficients of –3.359 for JLXooF/R,–3.426 for JLXocF/R,and –3.245 for JLBgF/R,corresponding to PCR efficiencies of 102.7%,95.8%,and 107.9%,respectively(Figs.2-A,3-A,4-A).Melting curve analysis showed a single peak for each primer at around 85 °C (Figs.2-B,3-B,4-B)suggesting the absence of primer dimers.The cycle threshold(Ct)in a real-time PCR assay is defined as the number of cycles required for the fluorescent signal to pass the threshold.The sample is considered to be negative or to represent environmental contamination when the Ct value is above 38.5.The detection limits of the genomic DNAs by SYBR Green PCR were 1 fg μL-1for OS198(Fig.2-C),1 fg μL-1for AHB4-75(Fig.3-C),and 10 fg μL-1for LMG2196(Fig.4-C).The primer sets developed in this study can be used to detect the presence of the target pathogens by both conventional and real-time PCR.

Fig.3-Sensitivity assay of JLXocF/R primer set for X.oryzae pv.oryzicola using SYBR Green RT-PCR.A:Standard curve.For each assay,templates(1-7)were diluted 10-fold to concentrations ranging from 1.0 to 1.0 × 10-6 ng μL-1.B:Melting-peak analysis.C:Fluorescence intensity;1.0 to 1.0 × 10-6 ng μL-1;1-7:samples;8:negative control.The arrow points to the limiting detection concentration of the primer set;D:CT(cycle threshold)and SE (standard error).
3.3.Multiplex PCR for detection of three pathogens and its sensitivity
To test further whether the primer sets could be used to detect the three target bacterial organisms simultaneously,artificial genomic DNA mixtures of OS198,AHB4-75,and LMG2196 were prepared based on different concentrations displayed in Table 3.When mix 1–4 was used as template in multiplex PCRs,all of the products specific to the three pathogens were visible on the 1.5%agarose gel(Table 3 and Fig.5).However,the specific amplicon of B.glumae was not detectable when mix 5 was used as template.Only the amplicon of X.oryzae pv.oryzicola was detected when mix 6 was used as template in multiplex PCR.The detection limits for the multiplex PCR assay were 0.3 pg μL-1for X.oryzae pv.oryzae,0.167 pg μL-1for X.oryzae pv.oryzicola,and 16.7 pg μL-1for B.glumae in the 20 μL reaction.The detection limits of each pathogen in multiplex PCR were highly similar to those of the single pathogen in conventional PCR.

Fig.4-Sensitivity assay of JLBgF/R primer set for B.glumae using SYBR Green RT-PCR.A:Standard curve.For each assay,templates(1-7)were diluted 10-fold to concentrations ranging from 1.0 to 1.0 × 10-6 ng μL-1.B:Melting-peak analysis.C:Fluorescence intensity;1.0 to 1.0 × 10-6 ng μL-1;1-7:samples;8:negative control.The arrow points to the limiting detection concentration of the primer set;D:CT(cycle threshold)and SE(standard error).
3.4.Pathogen detection in the artificial inoculated rice seeds
To determine whether multiplex PCR could detect the target pathogens in infected rice seeds,rice seeds were artificially infected by X.oryzae pv.oryzae,X.oryzae pv.oryzicola,or B.glumae and the mixture of the these three pathogens,respectively.If the seeds were infected by one pathogen,only the corresponding PCR product appeared on the gel using multiplex PCR assays.As a negative control,no amplification was observed from sterile distilled water-treated seeds.When the seeds were infected with a mixture of the three pathogens,the 230,164,and 112 bp fragments for X.oryzae pv.oryzae,X.oryzae pv.oryzicola,and B.glumae,respectively,were detected(Fig.6).

Table 3-Sample mixtures for multiplex PCR.

Fig.5-One-tube multiplex PCR for diagnosing three pathogens and its sensitivity.Lane M,DNA ladder(DL2000;TaKaRa);lanes 1-6 mixture of X.oryzae pv.oryzae strain OS225,X.oryzae pv.oryzicola AHB4-75,and B.glumae strain LMG2196,in concentrations 1 ng μL-1,5 × 10-1 ng μL-1,10 × 10-1 ng μL-1,5 × 10-2 ng μL-1,1 × 10-2 ng μL-1,5 × 10-3 ng μL-1.
4.Discussion
Conventionally,identification or detection of a plant pathogen requires pathogen isolation,cultivation,and verification based on bacteriological characteristics,colony morphology,electron microscopic observation,and other means–a timeconsuming process.In addition,the detection process requires much equipment and chemicals,increasing the cost.In the present study,an efficient multiplex PCR method was used to rapidly and accurately detect the rice bacterial pathogens X.oryzae pv.oryzae,X.oryzae pv.oryzicola,and B.glumae simultaneously in infected rice seeds,using new specific primer sets developed from specific sequence comparisons of X.oryzae pv.oryzae,X.oryzae pv.oryzicola,and B.glumae against their closely related species.
The bottleneck for PCR-based diagnostic or detection tools has been the availability of pathogen-specific primers.Sequence polymorphisms of 16S–23S ITS are often observed in strains of different species.In previous studies,specific DNA primers and probes have been designed based from 16S–23S ITS sequences for identification,separation and classification of some species of pathogens [6,9,17,27–32].16S–23S ITS of different species of Burkholderia were used to separate B.glumae from other Burkholderia species.However,it is difficult to separate pathovars using 16S–23S ITS [9].With advances in sequencing techniques,more and more bacterial genomic DNA sequences have been deposited in the GenBank database,allowing the development of specific primers using genomic comparisons[21].By genomic comparison among the X.oryzae pv.oryzae strains (PXO99A,MAFF311018,and KACC 10331),X.oryzae pv.oryzicola strains(BLS256),we identified the putative glycosyltransferase gene specific to X.oryzae pv.oryzae,and the AvrRxo gene specific to X.oryzae pv.Oryzicola (X.Wang,unpublished data).We then designed specific primers from the polymorphic DNA regions of these specific genes (Figs.S1,S2,S3).Although we used a limited number of strains of each pathogen,the primer sets we developed were specific.We amplified no sequences from the closely related bacterial pathogens X.campestris,X.maltophilia,B.gladioli pv.alliicola,or B.cepacia,or from the fungal pathogens,M.oryzae and U.oryzae.

Fig.6-Pathogen detection in artificial inoculated rice seeds.One-tube multiplex PCR for diagnosing three pathogens.Lane M,DNA ladder(DL 2000;TaKaRa);lane 1:seeds infected by X.oryzae pv.oryzae strain OS198;lane 2:seeds infected by X.oryzae pv.oryzicola strain AHB4-75;lane 3:seeds infected by B.glumae strain LMG;lane 4:mixture of seeds infected by OS198,AHB4-75,and LMG;lane 5:negative control.
For pathogen quarantine and inspection,primer sets are often required to be not only specific to the templates,but also sensitive to small quantities of the pathogens.Given that the amplified PCR fragments ranged from 112 to 230 bp in length,these primer sets can be used for both conventional and SYBR Green PCR.This knowledge will allow users to select the desired PCR platform to detect the pathogens.
Multiplex PCR has been applied to detect several pathogens in one PCR tube.Given that the lengths of the amplicons were very different,they were clearly visible on the 1.5%agarose gel after 50 min of separation.When complex templates consisting of three mixed samples were used,the detection limits of each sample were highly similar to those when single samples was used as the PCR template,suggesting that the multiplex PCR developed in the study can be used for simultaneous detection of the three rice bacterial pathogens.One common problem is that the detection sensitivity of multiplex PCR is lower than that of real-time PCR.To determine whether each primer set could amplify the corresponding DNA fragment from mixed samples with multiple pathogens using SYBR Green real time PCR,we made the following DNA mixtures: 1.DNA of OS198,AHB4-75 and LMG2196 with 1 ng μL-1at equal volume;and 2.detection limits of OS198,AHB4-75,and LMG2196 at equal volume.We observed specific real-time PCR products using the complex genomic DNA as templates and with even tiny amounts of DNA(Fig.S7).These findings suggest that our primers are specific and sensitive for simultaneous use in both multiplex and real-time PCR.
Sowing rice seeds containing the organisms of X.oryzae pv.oryzae,X.oryzae pv.oryzicola,or B.glumae can cause severe yield and economic losses in rice production.Rice leaves naturally infected by X.oryzae pv.oryzae and X.oryzae pv.oryzicola were collected from rice fields in Hangzhou in 2013 and infections were verified by phenotypic examination.The mixture of primer sets was used to detect different pathogens in these diseased leaves using multiplex PCR.The PCR products expected from positive controls were amplified using DNA from diseased leaf tissue infected by X.oryzae pv.oryzae and X.oryzae pv.oryzicola (Fig.S8),suggesting that these primer sets are highly effective and specific.
In conclusion,we have developed a user-friendly PCR based method to detect pathogens at extremely low levels in infected rice seeds and leaves.This method should be tested using diseased rice seeds from commercial fields before worldwide adoption for rapid pathogen inspection and quarantine.
We thank Professor Guanlin Xie of Zhejiang University for supplying B.glumae strain,Dr.Zhen Zhang of Zhejiang Academy of Agricultural Sciences for supplying the strains of X.oryzae pv.oryzae and X.oryzae pv.oryzicola,Dr.Yuan Fang of Zhejiang Normal University for supplying B.gladioli pv.alliicola strain and B.cepacia strain,and Dr.Stefano Costanzo of USDA APHIS-PPQ and Tracy Bianco of USDA-ARS DB NRRC for the critical review.This work was performed with the support of the National 863 Project (2012AA021601) and the New Seedling program for graduate students of Zhejiang Province(2012R409012).USDA is an equal opportunity provider and employer.
Supplementary material
Supplementary material related to this article can be found online at http://dx.doi.org/10.1016/j.cj.2014.06.005.
[1] T.W.Mew,Current status and future prospects of research on bacterial blight of rice,Annu.Rev.Phytopathol.25(1987)359–382.
[2] T.W.Mew,A.M.Alvarez,J.E.Leach,J.Swings,Focus on bacterial blight of rice,Plant Dis.77(1993) 5–12.
[3] M.Goto,Fundamentals of Bacterial Plant Pathology,Academic Press,San Diego,CA,1992.210–224.
[4] S.H.Ou,Rice Diseases,2nd edn Commonwealth Mycological Institute,Kew,Surrey,England,1985.380.
[5] A.P.K.Reddy,K.Krishnaiah,Z.T.Zhang,Y.Shen,Managing vulnerability of hybrid rice to biotic stresses in China and India,in:S.S.Virmani,E.A.Siddiq,K.Muralidharan (Eds.),Proceedings of the 3rd International Symposium on Hybrid Rice Technology: Advances in Hybrid Rice Technology,Hyderabad,India &International Rice Research Institute,Philippines,1998,pp.147–156.
[6] K.Goto,K.Ohata,New bacterial disease of rice(brown stripe and grain rot),Ann.Phytopathol.Soc.Jpn.21(1956) 46–47.
[7] R.Nandakumar,A.K.M.Shahjahan,X.L.Yuan,E.R.Dickstein,D.E.Groth,C.A.Clark,R.D.Cartwright,M.C.Rush,Burkholderia glumae and B.gladioli cause bacterial panicle blight in rice in the southern United States,Plant Dis.93(2009)896–905.
[8] J.H.Ham,R.A.Melanson,M.C.Rush,Burkholderia glumae:next major pathogen of rice? Mol.Plant Pathol.12(2011)329–339.
[9] N.Adachi,T.Oku,PCR-mediated detection of Xanthomonas oryzae pv.oryzae by amplification of the 16S–23S rDNA spacer region sequence,J.Gen.Plant Pathol.66(2000) 303–309.
[10] N.Sakthivel,C.N.Mortensen,S.B.Mathur,Detection of Xanthomonas oryzae pv.oryzae in artificially inoculated and naturally infected rice seeds and plants by molecular techniques,Appl.Microbiol.Biotechnol.56(2001)435–441.
[11] C.M.Vera Cruz,L.Halda-Alija,F.J.Louws,D.Z.Skinner,M.L.George,R.J.Nelson,F.J.DeBruijn,C.W.Rice,J.E.Leach,Repetitive sequence-based polymerase chain reaction of Xanthomonas oryzae pv.oryzae and Pseudomonas species,Int.Rice Res.Notes 20 (1995) 23–24.
[12] M.S.Cho,M.J.Kang,C.K.Kim,Y.J.Seol,J.H.Hahn,S.C.Park,D.S.Park,Sensitive and specific detection of Xanthomonas oryzae pv.oryzae by real-time bio-PCR using pathovar-specific primers based on an rhs family gene,Plant Dis.95(2011)589–594.
[13] W.J.Zhao,S.Zhu,X.L.Liao,H.Chen,T.W.Tan,Detection of Xanthomonas oryzae pv.oryzae in seeds using a specific TaqMan probe,Mol.Biotechnol.35 (2007) 119–127.
[14] M.J.Kang,M.H.Kim,D.J.Hwang,M.S.Cho,Y.Seol,J.H.Hahn,D.S.Park,Quantitative in planta PCR assay for specific detection of Xanthomonas oryzae pv.oryzicola using putative membrane protein based primer set,Crop.Prot.40 (2012)22–27.
[15] M.J.Kang,J.K.Shim,M.S.Cho,Y.Seol,J.H.Hahn,D.J.Hwang,D.S.Park,Specific detection of Xanthomonas oryzae pv.oryzicola in infected rice plant by use of PCR assay targeting a membrane fusion protein gene,J.Microb.Biotechnol.18(2008) 1492–1995.
[16] H.Zhang,Y.H.Jiang,B.S.Hu,F.Q.Liu,Z.G.Xu,Specific detection of Xanthomonas oryzae pv.oryzicola by PCR techniques,Acta Phytopathol.Sin.38(2008) 1–5(in Chinese with English abstract).
[17] N.Furuya,U.R.A.Hiroyuki,K.Iiyama,M.Matsumoto,M.Takeshita,Y.Takanami,Specific oligonucleotide primers based on sequences of the 16S–23S rDNA spacer region for the detection of Burkholderia gladioli by PCR,J.Gen.Plant Pathol.68(2002) 220–224.
[18] Y.Maeda,H.Shinohara,A.Kiba,K.Ohnishi,N.Furuya,Y.Kawamura,Y.Hikichi,Phylogenetic study and multiplex PCR-based detection of Burkholderia plantarii,Burkholderia glumae and Burkholderia gladioli using gyrB and rpoD sequences,Int.J.Syst.Evol.Microbiol.56(2006) 1031–1038.
[19] Y.Huai,L.H.Xu,S.H.Yu,G.L.Xie,Real-time fluorescence PCR method for detection of Burkholderia glumae from rice,Chin.J.Rice Sci.23(2009) 107–110 (in Chinese with English abstract).
[20] R.J.Sayler,R.D.Cartwright,Y.Yang,Genetic characterization and real-time PCR detection of Burkholderia glumae,a newly emerging bacterial pathogen of rice in the United States,Plant Dis.90(2006) 603–610.
[21] J.M.Lang,J.P.Hamilton,M.G.Q.Diaz,M.A.Van Sluys,M.R.G.Burgos,C.M.Vera Cruz,J.E.Leach,Genomics-based diagnostic marker development for Xanthomonas oryzae pv.oryzae and X.oryzae pv.Oryzicola,Plant Dis.94(2010)311–319.
[22] X.L.Liao,S.F.Zhu,W.J.Zhao,K.Luo,Y.X.Qi,Detection and identification of Xanthomonas oryzae pv.oryzae and Xanthomonas oryzae pv.oryzicola by real-time fluorescent PCR,Acta Microbiol.Sin.43(2003) 626–634.
[23] N.J.Talbot,D.J.Ebbole,J.E.Hamer,Identification and characterization of MPG1,a gene involved in pathogenicity from the rice blast fungus Magnaporthe grisea,Plant Cell 5(1993) 1575–1590.
[24] U.M.Csaikl,H.Bastian,R.Brettschneider,S.Gauch,A.Meir,M.Schauerte,B.Ziegenhagen,Comparative analysis of different DNA extraction protocols: a fast,universal maxi-preparation of high quality plant DNA for genetic evaluation and phylogenetic studies,Plant Mol.Biol.Rep.16(1998) 69–86.
[25] W.K.Kim,W.Mauthe,G.Hausner,G.R.Klassen,Isolation of high molecular weight DNA and double-stranded RNAs from fungi,Can.J.Bot.68(1990) 1898–1902.
[26] T.A.Hall,Bioedit: a user-friendly biological sequence alignment editor and analysis program for window 95/98/NT,Nucleic Acids Symp.Ser.41(1999) 95–98.
[27] J.Garcia-Martinez,S.G.Acinas,A.I.Anton,F.Rodriguez-Valera,Use of the 16S–23S ribosomal genes spacer region in studies of prokaryotic diversity,J.Microbiol.Methods 36(1999)55–64.
[28] J.García-Martínez,I.Bescós,J.J.Rodríguez-Sala,F.Rodríguez-Valera,RISSC:a novel database for ribosomal 16S–23S RNA genes spacer regions,Nucleic Acids Res.29(2001) 178–180.
[29] E.R.Gonçalves,Y.B.Rosato,Phylogenetic analysis of Xanthomonas species based upon 16S–23S rDNA intergenic spacer sequences,Int.J.Syst.Evol.Microbiol.52(2002)355–361.
[30] V.Gürtler,V.A.Stanisich,New approaches to typing and identification of bacteria using the 16S–23S rDNA spacer region,Microbiology 142 (1996) 3–16.
[31] L.Hauben,L.Vauterin,J.Swings,E.R.B.Moore,Comparison of 16S ribosomal DNA sequences of all Xanthomonas species,Int.J.Syst.Bacteriol.47(1997) 328–335.
[32] A.Roth,M.Fischer,M.E.Hamid,S.Michalke,W.Ludwig,H.Mauch,Differentiation of phylogenetically related slowly growing mycobacteria based on 16S–23S rRNA gene internal transcribed spacer sequences,J.Clin.Microbiol.36 (1998)139–147.

- The Crop Journal的其它文章
- Maize forage aptitude: Combining ability of inbred lines and stability of hybrids
- Growth,photosynthesis and nitrogen metabolism in soybean varieties after exclusion of the UV-B and UV-A/B components of solar radiation
- Differences between soybean genotypes in physiological response to sequential soil drying and rewetting
- Mapping and validation of a dominant salt tolerance gene in the cultivated soybean(Glycine max) variety Tiefeng 8
- Genetic background effects on QTL and QTL × environment interaction for yield and its component traits as revealed by reciprocal introgression lines in rice
- Brief Guide for Authors