
NLRP3 炎性小体在中枢神经系统炎症中的作用
2014-03-11 08:20宋丽敏综述审校
中风与神经疾病杂志 2014年4期
宋丽敏 综述,尚 游,吴 艳 审校
固有免疫是机体重要的防御屏障,含有核苷酸结合结构域和富含亮氨酸重复序列的受体(nucleotide-binding domain leucine-rich repeat-containing receptors,NLR)是一种模式识别受体,在机体受到微生物感染和内源性损伤时,可以识别病原相关分子模式和损伤相关分子模式并被激活,形成炎性小体,激活caspase-1,参与固有免疫应答。
1 炎性小体
炎性小体是细胞内的蛋白质复合体,主要存在于固有免疫细胞中。在外周主要是巨噬细胞、树突状细胞;在中枢神经系统则主要是小胶质细胞。目前已经发现了多种炎性小体复合物,典型组成包括NLR、接头分子含有CARD 的凋亡相关斑点样蛋白(apoptosis-associated speck-like protein containing CARD,ASC)和效应蛋白caspase-1 前体(pro-caspase-1)(见图1)。
NLRs 是胞浆内的免疫受体家族,在人类细胞中已经发现了超过20 种成员。NLRs N 端差异比较大,因此可以据此对NLR 进行分类,已经了解的N 端结构域有4 种:N 端由pyrin domain(PYD)组成的称为NLRP;N 端由caspase 募集结构域(CARD)组成的称为NLRC;N 端由BIR 样结构域组成的称为NAIP 或BIRC;另一种是酸性反式激活结构域。C 端是亮氨酸重复序列(leucine-rich repeats,LRR),与配体识别有关,在NLR 自身调节方面也有作用。N 端与C 端之间的是核苷酸结合结构域(nucleotide-binding and oligomerization domain,NBT),与自身寡聚化有关。ASC 由PYD 和CARD 组成,有些NLR 不能直接与pro-caspase 结合,ASC 可以起到衔接作用。刺激因素的作用下,NLRP3 寡聚化,通过PYD 结构域与ASC 结合,ASC 再通过CARD 结构域与pro-caspase-1 同源结合,从而形成多蛋白炎性小体。Pro-caspase-1 在胞浆组成性表达,炎性小体组装形成以后,通过调节两个或多个pro-caspase-1 单体的空间位置关系,启动caspase-1 自身催化活性,使之切割为p20 和p10 两个亚基,即成为成熟的有活性的caspase-1。活化的caspase-1 切割IL-1β 的前体pro-IL-1β,使之成熟并分泌到细胞外发挥生物学效应。NLRP3 与ASC 和pro-caspase-1 组装形成的NLRP3 炎性小体是本文的主要讨论对象。
2 NLRP3 炎性小体
2.1 NLRP3 炎性小体的激活物 有活性的NLRP3 炎性小体的产生需要两个信号:信号一是炎性刺激通过NF-κB依赖性信号通路,引起NLRP3 表达;信号二是NLRP3 激活。NLRP3 可以由外源性微生物及其毒素和内源性危险信号激活,它们的理化特性各不相同。外源性的包括金黄色葡萄球菌及其溶血毒素[1]、李斯特单核杆菌、结核分枝杆菌[2]、肺炎链球菌[3]、腺病毒等。内源性的包括三磷酸腺苷(ATP)、石棉、二氧化硅、尿酸盐、7-溴靛玉红-3、神经酰胺、脂质体、氧化低密度脂蛋白[4]等。
2.2 NLRP3 炎性小体的激活机制 没有内外源性刺激因素作用时,NLRP3 的LRR 结构域通过与泛素连接酶相关蛋白和分子伴侣热休克蛋白90 结合,处于自身抑制状态,但是易于被激活[1]。NLRP3 炎性小体活化的确切机制目前还没有完全明确,目前,被大家较多提及的理论有以下这些(见图2)。
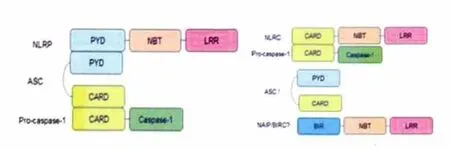
图1 炎性小体的基本组成

图2 NLRP3 炎性小体的激活途径:(1)钾离子外流;(2)ROS产生;(3)吞噬溶酶体内容物释放;(4)钙离子动员
2.2.1 钾离子外流 许多刺激物如微生物的成孔毒素、结晶体,能在细胞膜上形成孔道,引起钾离子外流[5]。细胞死亡、脑缺血等因素都会引起ATP 释放到胞外,ATP 激活胞膜的P2X7 受体,打开该配体门控的离子通道,使钾离子外流。P2X7 受体的激活还可以引起细胞膜上名为泛连接蛋白的大孔道开放,能转运900 道尔顿大小的分子穿过细胞膜,包括钾离子[1,6]。在培养基中加入高浓度氯化钾,抑制钾离子外流,发现结核分枝杆菌等诱导的IL-1β 的产生明显减少[2]。近来,ATP 释放和嘌呤受体信号在NLRP3 炎性小体活化中的作用机制已经得到较为清楚的阐释。
2.2.2 活性氧 活性氧物质(reactive oxygen species,ROS)与氧化应激有关,细胞接受刺激时,氧化酶组装活化,线粒体内ROS 合成[7],是NLRP3 炎性体活化的重要上游信号。胞浆内硫氧还蛋白结合蛋白(TXNIP)与硫氧还蛋白(TRX)结合,炎性小体的激活物使ROS 产生增加,TXNIP 与TRX 解离,TXNIP 构象改变,与NLRP3 相互作用,引起炎性小体活化[8]。抑制ROS 产生的药物或者线粒体ROS 清除剂都能减少IL-1β 产生[9]。但是,也有研究者认为,ROS 通过NF-κB 途径上调NLRP3 和pro-IL-1β 的表达,而不是直接活化NLRP3 炎性小体。为了特异性地研究ROS 是否在pro-IL-1β 合成以后对NLRP3 炎性小体有影响,在脂质体激活NLRP3 炎性小体模型中,延长脂多糖提前处理的时间,以保证NF-κB 途径的充分活化,再加入抑制ROS 产生的药物,结果同样证实ROS 可以活化NLRP3 炎性小体[4]。
2.2.3 溶酶体内容物释放 溶酶体内有多种蛋白水解酶,包括组织蛋白酶家族,其破坏后这些蛋白水解酶释放入胞浆,可以引起损伤作用,组织蛋白酶B(Cathepsin B)与阿尔兹海默病(Alzheimer disease,AD)有关。Aβ 被小胶质细胞吞噬后,溶酶体肿胀、不稳定、功能失常,Cathepsin B 释放入胞浆,引起NLRP3 炎性小体活化,Cathepsin B 抑制剂使IL-1β释放明显减少[10]。血清淀粉样蛋白A、7-溴靛玉红-3、金黄色葡萄球菌引起NLRP3 炎性小体活化时也是如此。结核分枝杆菌激活NLRP3 炎性小体时,除了Cathepsin B,组织蛋白酶L 也发挥作用[5]。P2X7 受体激活也可引起溶酶体内容物释放。肺炎链球菌引起ATP 释放到胞外,继而溶酶体组织蛋白酶B 释放,NLRP3 炎性小体激活,但是没有巨噬细胞吞噬和溶酶体破坏的参与[3]。
2.2.4 其他 钙离子动员在NLRP3 炎性小体活化中的作用也逐渐被人发现。细胞接受刺激时发生钙离子动员,钾离子外流引起浆膜极化,钙离子内流,吞噬溶酶体破裂也可以引起其内的钙离子进入胞浆。一方面,胞浆内钙超载促进线粒体损伤,产生更多ROS,进一步活化NLRP3;另一方面,单核巨噬细胞浆膜上表达瞬时受体电位通道2,是一种非选择性阳离子通道,ROS 在胞浆的聚集可以引起钙离子通过此通道内流。该通道基因敲除小鼠巨噬细胞中,钙离子内流减少,IL-1β 释放也减少,提示经此途径引起的钙离子内流与NLRP3 炎性小体活化有关[11]。抑制钙离子动员可以抑制NLRP3 炎性小体活化。
以上这些学说都有待阐明,越来越多人接受的观点是,NLRP3 炎性小体的激活是个复杂的、多种机制相互作用、相互影响的过程。
2.3 NLRP3 炎性小体的活性调节
2.3.1 ASC 调节 细胞未接受刺激时,ASC 位于胞核,炎性刺激使ASC 转位到胞浆才能与NLRs 和pro-caspase-1 结合成为聚合体,进而引起IL-1β 处理和分泌。ASC 有3 种新的亚型:ASC-b、ASC-c 和ASC-d,它们的功能各不相同。只有活化状态的ASC 和ASC-b 能与NLRP3 和caspase-1 共定位,激活NLRP3 炎性小体。ASC-c 和ASC-d 作为抑制亚基,前者只与caspase-1 共定位,后者既不与NLRP3 也不与caspase-1共定位,都不发挥活化作用[12]。
2.3.2 特殊蛋白调节 COPs(CARD-only proteins)是只含有CARD 结构域的蛋白家族,目前发现的成员包括Iceberg、COP1/Pseudo-ICE、INCA、caspase-12s、和Nod2-S。它们与pro-caspase-1 的CARD 结构域结合,使后者不能与ASC 结合,不能形成炎性小体。POPs(PYD-only proteins)是只含有PYD 结构域的蛋白家族,较多提及的是POP1 和POP2,与ASC 或者NLRP 的PYD 结构域结合,阻止炎性小体形成;还可以阻止NF-κB 的激活,从转录水平抑制NLRP3 表达。一些病毒株可以通过自身的病毒POPs 抑制炎性小体活化[14]。
2.3.3 其他调节 细胞间相互作用也可以调控炎性小体的激活。小鼠CD4+效应T 细胞和记忆B 细胞能够抑制caspase-1 活化和IL-1β 成熟,主要是针对NLRP3 和NLRP1,通过T 细胞表达的肿瘤坏死因子家族配体实现[13]。细胞自噬对NLRP3 炎性小体活化也有调节作用。自噬过程调控长寿命蛋白质、蛋白多聚物和入侵微生物的降解,缺乏自噬功能的细胞,NLRP3 炎性小体更易被激活[9,14]。
3 NLRP3 炎性小体与中枢神经系统炎症
小胶质细胞是在中枢神经系统定居的免疫细胞,其活化可以引起炎症反应。正常情况下,IL-1β 在脑中有低水平表达,NLRP3 炎性小体激活使IL-1β 大量成熟并分泌到细胞外,引起炎症反应,激活一些病理生理过程。多种中枢神经系统炎性反应与NLRP3 炎性小体的活化有关。
3.1 NLRP3 炎性小体与神经退行性疾病 细胞外Aβ聚集形成的斑块在AD 的发生发展中有重要作用,小胶质细胞募集在Aβ 斑块周围,发挥吞噬作用并被活化,IL-1β 是针对Aβ 的炎症反应的主要细胞因子,AD 患者脑脊液中IL-1β含量明显升高。Aβ 被小胶质细胞吞噬后激活NLRP3 炎性小体,M2 型标记物Arg-1、IL-4 表达减少,M1 型标记物NOS2等表达增多,说明小胶质细胞从M2 型转变为M1 型[15]。产生的IL-1β 和其他的促炎性细胞因子、神经毒性因子,对周围的神经元等造成损伤,进一步放大Aβ 的致病作用[10]。同时,活化的小胶质细胞清除Aβ 的能力下降,引起Aβ 斑块沉积增加。
多发性硬化症(MS)是中枢神经系统的自身免疫性炎性脱髓鞘疾病,MS 斑块和MS 患者血液中caspase-1 含量都有明显增高,脑脊液中IL-1β 明显增多,Nlrp3 基因变异的患者体内检测到MS 样损伤。实验性自身免疫性脑脊髓炎是MS的动物模型,该模型脱髓鞘病灶内有小胶质细胞的活化和聚集,而且早于临床症状出现,NLRP3 炎性小体活化可以加重病情进展[16]。
3.2 NLRP3 炎性小体与感染性疾病 脑脓肿的主要病因是金黄色葡萄球菌感染中枢神经系统,感染后数小时内就有小胶质细胞的激活,以及多种促炎性因子的快速产生,引起广泛的炎症和坏死。IL-1β 是金葡菌激活小胶质细胞产生的主要细胞因子,部分是通过激活NLRP3 炎性小体产生的,还有一部分是通过非炎性小体途径产生的。NLRP3 炎性小体的激活有赖于活细菌产生的ATP 和α-、γ-溶血毒素,cathepsin B 在此过程中也发挥作用[1]。
脑膜炎是肺炎链球菌感染性疾病中预后最差的,死亡率达到15%~30%,caspase-1 是该病炎症反应的主要调节物质。NLRP3 和ASC 表达缺失可以减低小鼠肺炎球菌性脑膜炎的临床评分、组织损伤程度和脑的炎症,用IL-1β 抑制剂可以观察到ASC 和NLRP3 依赖性的病理学缓解[3]。
小胶质细胞是结核杆菌在中枢神经系统感染的主要靶细胞,活化的小胶质细胞与其他细胞共同作用,释放多种促炎因子和趋化因子,结核性脑膜炎患者脑脊液中IL-1β 有明显增高。用结核分枝杆菌感染巨噬细胞,获得条件培养基,再用此处理小胶质细胞,最后经结核杆菌感染,可以发现明显的caspase-1 活化和IL-1β 释放,并且发现IL-1β 的成熟是NLRP3、ASC 和ROS 依赖性的。地塞米松治疗结核性脑膜炎的作用,部分是通过抑制ROS,从而抑制NLRP3 炎性小体激活来实现的[2]。
4 总结
NLRP3 炎性小体是近年来的研究热点,目前已经发现了多种内源性和外源性物质可以发挥激活作用,有关其活化和调节的分子机制也在逐渐阐明,NLRP3 炎性小体的激活引起IL-1β 成熟,在中枢神经系统固有免疫中发挥重要作用。特异性抑制NLRP3 炎性小体的活化途径,是相关疾病新的治疗靶点。
[1]Hanamsagar R,Torres V,Kielian T.Inflammasome activation and Il-1β/IL-18 processing are influenced by distinct pathways in miacroglia[J].J Neurochem,2011,119(4):736-748.
[2]Lee HM,Kang J,Lee SJ,et al.Microglial activation of the NLRP3 Inflammasome by the priming signals derived from macrophages infected with mycobacteria[J].Glia,2013,61(3):441-452.
[3]Hoegen T,Tremel N,Klein M,et al.The NLRP3 inflammasome contributes to brain injury in pneumococcal meningitis and is activated through ATP-dependent lysosomal cathepsin B release[J].J Immu-nol,2011,187(10):5440-5451.
[4]Lin J,Shou X,Mao X,et al.Oxidized low density lipoprotein induced caspase-1 mediated pyroptotic cell death in macrophages:implication in lesion instability[J].PLoS One,2013,8(4):62148.
[5]Fernandes-Alnemri T,Wu J,Yu JW,et al.The pyroptosome:a supramolecular assem bly of ASC dimers mediating inflammatory cell death via caspase-1 activation[J].Cell Death Differ,2007,14(9):1590-1604.
[6]Niemi K,Teirila L,Lappalainen J,et al.Serum Amyloid A activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B-sensitive pathway[J].J Immunol,2011,186(11):6119-6128.
[7]Dostert C,Perilli V,Van Bruggen R,et al.Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica[J].Science,2008,320(5876):674-677.
[8]Zhou R,Yazdi AS,Menu P,et al.A role for mitochondria in NLRP3 inflammasome activation[J].Nature,2011,469(7329):221-225.
[9]Nakahira K,Haspel JA,Rathinam VA,et al.Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome[J].Nat Immunol,2011,12(3):222-230.
[10]Halle A,Hornung V,Petzold GC,et al.The NALP3 inflammasome is involved in the innate immune response to amyloid-β[J].Nat Immunol,2008,9(8):857-865.
[11]Zhong Z,Zhai Y,Liang S,et al.TRPM2 links oxidative stress to NLRP3 inflammasome activation[J].Nat Commun,2013,4:1611.
[12]Bryan NB,Dorfleutner A,Kramer SJ,et al.Differential splicing of the apoptosis-associated speck like protein containing a caspase recruitment domain(ASC)regulates inflammasomes[J].J Inflamm(Lond),2010,7:23.
[13]Guarda G,Dostert C,Staehli F,et al.T cells dampen innate immune responses through inhibition of NLRP1 and NLRP3 inflammasomes[J].Nature,2009,460(7252):269-273.
[14]Saitoh TI,Fujita N,Jang MH,et al.Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production[J].Nature,2008,456(7219):264-268.
[15]Heneka MT,Kummer MP,Stutz A,et al.NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice[J].Nature,2013,493(7434):674-678.
[16]Gris DI,Ye Z,Iocca HA,et al.NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses[J].J Immunol,2010,185(2):974-981.
猜你喜欢
中西医结合心脑血管病杂志(2022年19期)2022-11-19
中国临床解剖学杂志(2022年1期)2022-11-15
西部中医药(2022年9期)2022-09-28
检验医学与临床(2022年12期)2022-06-27
中国人兽共患病学报(2022年5期)2022-06-08
河南科技(2022年6期)2022-04-22
生物化工(2021年2期)2021-01-19
微生物与感染(2020年6期)2020-12-28
中国医学影像学杂志(2020年7期)2020-08-04
微生物与感染(2019年6期)2019-12-30
