
c-Kit激酶抑制剂的研究与开发
2014-03-08 06:46王佩倍庞俊霞赖宜生
药学进展 2014年7期
王佩倍,庞俊霞,赖宜生
(中国药科大学新药研究中心,江苏 南京 210009)
·综述与专论·
REVIEW AND MONOGRAPH
c-Kit激酶抑制剂的研究与开发
王佩倍,庞俊霞,赖宜生*
(中国药科大学新药研究中心,江苏 南京 210009)

c-Kit是典型的Ⅲ型受体酪氨酸激酶,在肿瘤的发生发展以及侵袭、迁移和复发过程中起着十分重要的作用,是目前肿瘤分子靶向治疗的热门靶标之一,其抑制剂也成为抗肿瘤药物研究与开发的热点。简介c-Kit激酶及其激活型突变与肿瘤发生发展的关系,着重综述近年来已上市和处于临床试验阶段的c-Kit激酶抑制剂及其耐药机制研究。
c-Kit激酶抑制剂;c-Kit激活型突变;c-Kit耐药型突变;肿瘤治疗
受体酪氨酸激酶c-Kit(又称CD117)是由逆转录病毒原癌基因c-kit编码的一类具有酪氨酸激酶活性的跨膜受体蛋白,与血小板衍生生长因子受体(PDGFR)、巨噬细胞集落刺激因子-1受体(CSF-1R)和Fms样酪氨酸激酶受体3(FLT3)共同组成Ⅲ型受体酪氨酸激酶超家族,其在肿瘤发生发展过程中起着十分重要的作用[1]。因此,c-Kit是目前肿瘤分子靶向治疗的热门靶标之一。
1 c-Kit激酶
编码c-Kit激酶的原癌基因c-kit位于人体4号染色体(4q11-12)上,长约90 kb,由21个外显子组成,其编码的c-Kit是相对分子质量为145 000的跨膜糖蛋白,全长为976个氨基酸,在进化过程中高度保守[2]。
c-Kit激酶由胞外域、跨膜域和胞内域组成,其胞外域位于N末端,包含520个氨基酸,由5个免疫球蛋白样结构域(D1~D5)组成,其中D1~D3是配体结合区,D4~D5是单体二聚化区;跨膜域包含23个氨基酸;胞内域即酪氨酸激酶结构域位于C末端,包含433个氨基酸,可细分为近膜区(JMD)、三磷酸腺苷(ATP)结合区、激酶插入区和A-loop区(见图1)[3]。
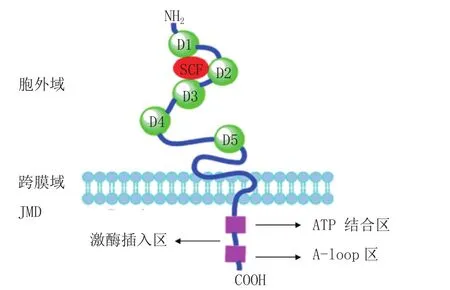
图1 c-Kit激酶的结构Figure 1 Structure of c-Kit kinase
目前已发现,c-Kit激酶通常呈现活化和非活化2种构象(见图2),二者之间存在一个动态平衡。当c-Kit无配体结合时,JMD处于激酶活性位点中,而A-loop区则堵住ATP进入激酶活性位点的路径,其N末端天冬氨酸-苯丙氨酸-甘氨酸(DFG)序列处于非催化状态,此时激酶的非活化构象占主导地位;当c-Kit与配体结合时,引发的二聚化导致JMD中Tyr568和Tyr570磷酸化,减弱JMD与激酶活性位点的相互作用,而A-loop区则离开活性位点,DFG也处于适合ATP结合和催化的位置,此时激酶的活化构象占主导地位[4-5]。简而言之,JMD能调节c-Kit的非活化构象,A-loop区则能调节c-Kit的活化构象。
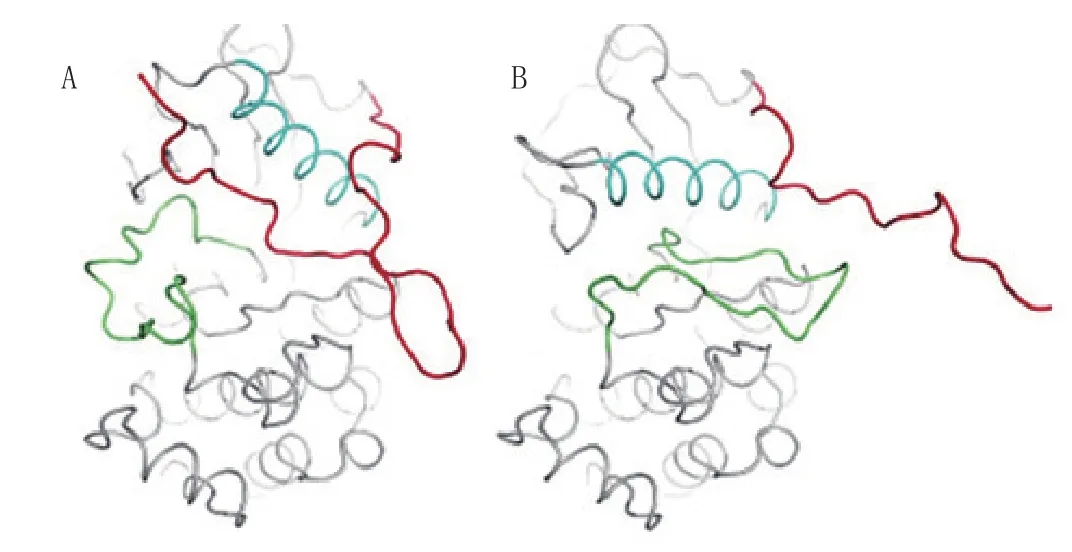
图2 c-Kit激酶的2种构象Figure 2 Two conformations of c-Kit kinase
2 c-Kit激活型突变与肿瘤的发生发展
c-Kit激酶被干细胞生长因子激活后,形成二聚体,导致JMD中Tyr568和Tyr570的跨膜磷酸化,从而改变JMD的三维结构,减弱其与激酶活性位点的相互作用,即减弱激酶的自抑制作用,致使激酶区磷酸化,进而募集下游信号分子,最终激活下游信号通路,调节细胞的生长和增殖。目前已知,c-Kit在多种肿瘤中处于异常活化状态,这主要源于c-Kit突变导致其过度表达和持续性激活[6]。c-Kit激活型突变最初是在人类肥大细胞白血病细胞系HMC-1中发现,即JMD的V560G突变和激酶区的D816V突变,这2种突变均能引起酪氨酸的自磷酸化,导致c-Kit的非配体依赖性激活(Furitsu等,J Clin Invest,1993年)。
常见的c-Kit激活型突变位点如图3所示[7],其主要发生在胞外域(外显子8、9)、胞内JMD(外显子11)以及激酶结构域中A-loop区(外显子17),这些突变均会破坏c-Kit的自抑制机制,从而导致其持续激活,促进肿瘤的发生发展[3]。值得一提的是,不同肿瘤的常见c-Kit突变位点存在差异。
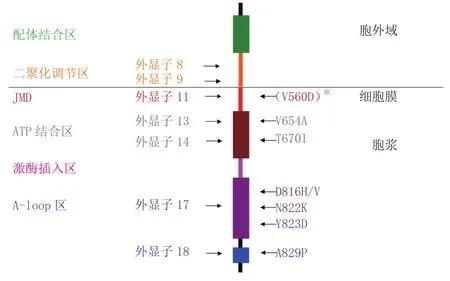
图3 常见c-Kit激活型突变位点的分布Figure 3 Common location of c-Kit activating mutations
在胃肠道间质瘤(GIST)中,约有80%存在c-Kit激活型突变[8],而这些突变主要发生在外显子11(如V560D),其次是外显子9(如一个AY插入)[9]。外显子11编码的JMD是受体酪氨酸激酶的关键自调节区域,能稳定激酶的非活化构象[4]。在GIST中,c-kit外显子11存在多种突变类型,包括点突变、串联重复、缺失和插入,其各自精确的机制可能有所不同,但结果都会阻止JMD与激酶活性位点的相互作用,导致c-Kit不依赖配体的持续活化[3]。
在黑色素瘤中,常见的c-Kit激活型突变发生于外显子11和13,如L576P和K642E[10]。在c-Kit的非活化构象中,642位赖氨酸与574位苏氨酸形成的氢键能稳定非活化构象,而576位亮氨酸不仅能稳定JMD中一小段螺旋结构,还能与α-C螺旋(Val643,Tyr646)形成氢键作用,因此Leu576和Lys642中的任何一个位点发生突变都会引起非活化构象的不稳定,从而导致c-Kit向活化构象转变。
在系统性肥大细胞增多症(SM)以及核结合因子相关的急性髓细胞白血病(AML)细胞中,c-Kit激活型突变主要发生在外显子17,如D816V/H/Y[11]。
3 c-Kit激酶抑制剂
c-Kit的激活型突变与多种肿瘤的发生发展密切相关,因此,它成为肿瘤治疗的一个有效靶标,其抑制剂也成为抗肿瘤药物研究与开发的热点。
3.1 已上市的c-Kit激酶抑制剂
在临床治疗中取得巨大成功的伊马替尼(格列卫TM)是首个上市的c-Kit激酶抑制剂,自其于2001年上市以来,目前已有9个c-Kit激酶抑制剂药物相继上市(见表1)。值得一提的是,由于大部分c-Kit激酶抑制剂均是ATP竞争型抑制剂,而不同类型的酪氨酸激酶之间又有较高的同源性,因此目前已上市的c-Kit激酶抑制剂均是多靶点抑制剂,即在抑制c-Kit的同时,还可抑制其他类型的酪氨酸激酶。
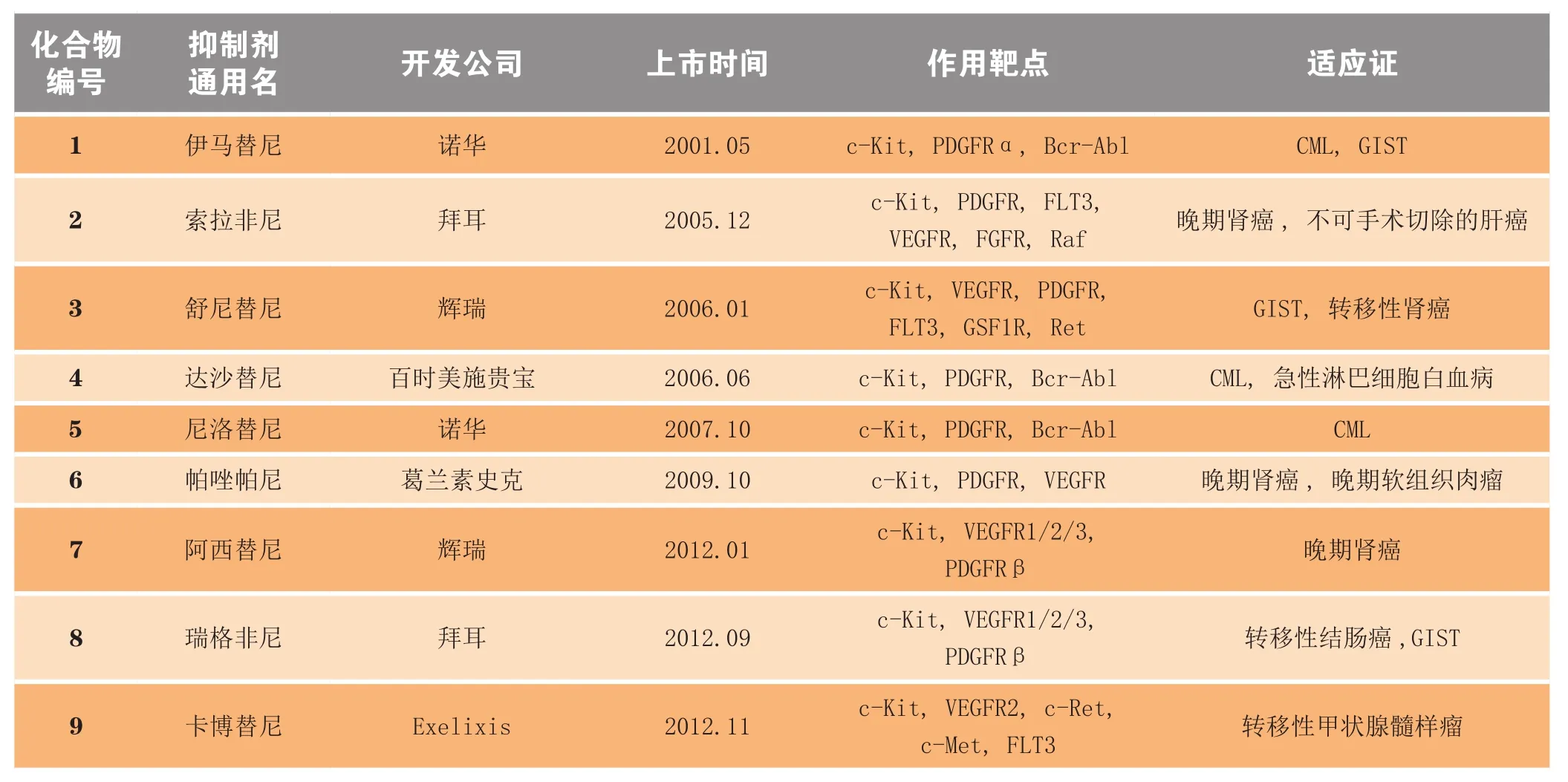
表1 已上市的c-Kit激酶抑制剂Table 1 Marketed c-Kit kinase inhibitors
伊马替尼(GleevecTM)是诺华公司开发的首个获美国FDA批准上市的酪氨酸激酶抑制剂,开启了肿瘤分子靶向治疗的先河,目前是国际公认的治疗CML和GIST的一线用药。然而,随着伊马替尼在临床上的广泛应用,其引发的肿瘤耐药性问题愈发严重。为此,百时美施贵宝公司和诺华公司分别于2006年和2007年推出了第2代酪氨酸激酶抑制剂达沙替尼(SprycelTM)和尼洛替尼(TasignaTM),用于治疗对包含伊马替尼在内的治疗方案耐药或不能耐受的CML患者。相关临床研究证明这2种酪氨酸激酶抑制剂在诱导产生主要分子学反应和完全细胞遗传学反应等方面的表现均优于伊马替尼,目前它们已成为CML治疗的一线药物[12-13]。
索拉非尼(NexavarTM)是拜耳公司开发的能同时作用于多种丝/苏氨酸激酶和酪氨酸激酶(如B-Raf和VEGFR等)的抑制剂,在申报上市时便被美国FDA授予“快速通道”审批资格,最终于2005年上市,用于治疗晚期肾癌,随后又在2007年获批成为首个用于治疗晚期肝癌的口服药物[14]。
舒尼替尼(SutentTM)是辉瑞公司开发的吲哚酮类多靶点酪氨酸激酶抑制剂,2006年1月被美国FDA批准用于治疗对标准疗法无响应或不能耐受的GIST和转移性肾细胞癌,成为首个同时获准用于治疗这2种类型癌症的抗肿瘤药物。
帕唑帕尼(VotrientTM)是葛兰素史克公司开发的用于治疗晚期肾癌的酪氨酸激酶抑制剂,临床研究表明,其用于一线治疗转移性肾细胞癌患者所产生的以无进展生存期(PFS)为指标的疗效不亚于舒尼替尼,且在安全性和改善患者生活质量方面更优于舒尼替尼[15]。此外,2013年美国临床肿瘤学会ASCO年会上发布的国际合作组AGO-OVAR16 Ⅲ期临床试验结果显示,帕唑帕尼可有效延缓晚期卵巢癌的进展,因此有可能成为晚期卵巢癌维持治疗的新选择。
阿西替尼(InlytaTM)是辉瑞公司开发的又一个酪氨酸激酶抑制剂,于2012年1月由美国FDA批准用于治疗使用其他抗癌药无效的晚期肾癌。一项Ⅲ期临床研究在比较阿西替尼和索拉非尼作为二线治疗方案对转移性肾细胞癌患者的疗效时发现,阿西替尼治疗组患者的PFS更长[16]。
瑞格非尼(StivargaTM)是拜耳公司继索拉非尼之后开发的新一代酪氨酸激酶抑制剂,于2012年9月被美国FDA批准上市,至今已获准用于治疗转移性结直肠癌以及不能手术切除且对伊马替尼和舒尼替尼治疗无应答的晚期GIST。一项旨在评估瑞格非尼作为二线治疗药物用于索拉非尼治疗失败的肝癌患者时的安全性和有效性的Ⅱ期临床试验表明,瑞格非尼具有可耐受的安全性,且抗肝癌疗效明显,有望作为中晚期肝癌患者的二线治疗药物[17]。2014年2月,拜耳公司宣布启动一项新的全球性瑞格非尼Ⅲ期临床试验,拟在北美、巴西、欧洲、亚洲、以色列及澳大利亚等地区和国家招募大约750名癌已扩散至肝脏的结肠直肠癌受试者,这项随机、双盲、安慰剂对照、名为COAST的临床研究旨在评价瑞格非尼辅助治疗肝脏肿瘤已被切除患者的效果[18]。
卡博替尼(CometriqTM)是Exelixis公司开发的一种作用于c-Kit、VEGFR2、c-Ret、c-Met和FLT3的多靶点抑制剂,在2012年11月被美国FDA批准用于治疗转移性甲状腺髓样瘤(MTC)。早在Ⅰ期临床试验中该药便显示出对MTC患者的疗效,随后意大利比萨大学的Rossella Elisei博士等人在此基础上进行了一项该药的双盲Ⅲ期临床试验,结果显示,该药以140 mg·d-1剂量用于出现恶化的转移性MTC 患者,可明显延长受试者的PFS,并具有统计学意义,表明其可用作这种罕见性疾病的一个重要的新治疗手段[19]。
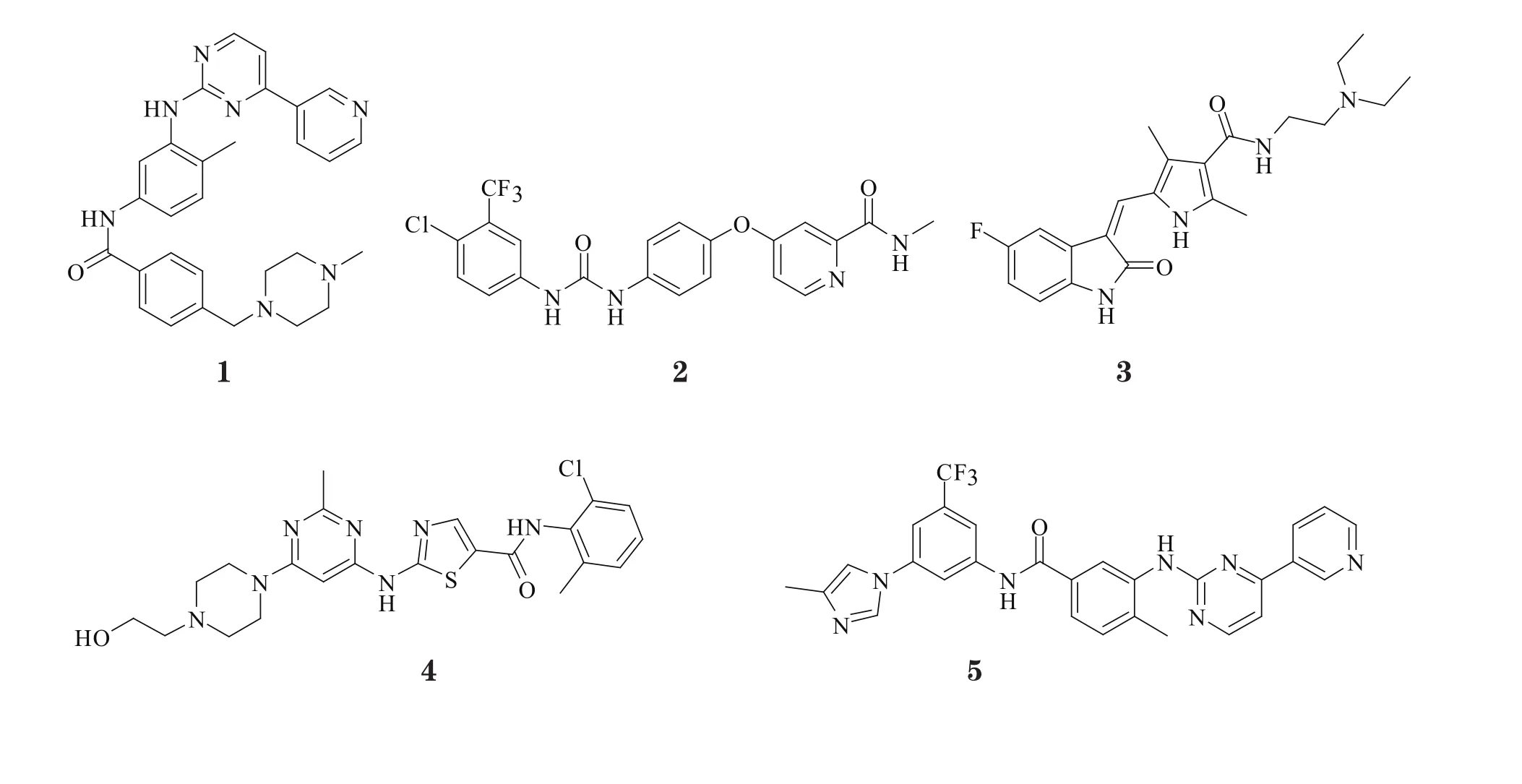
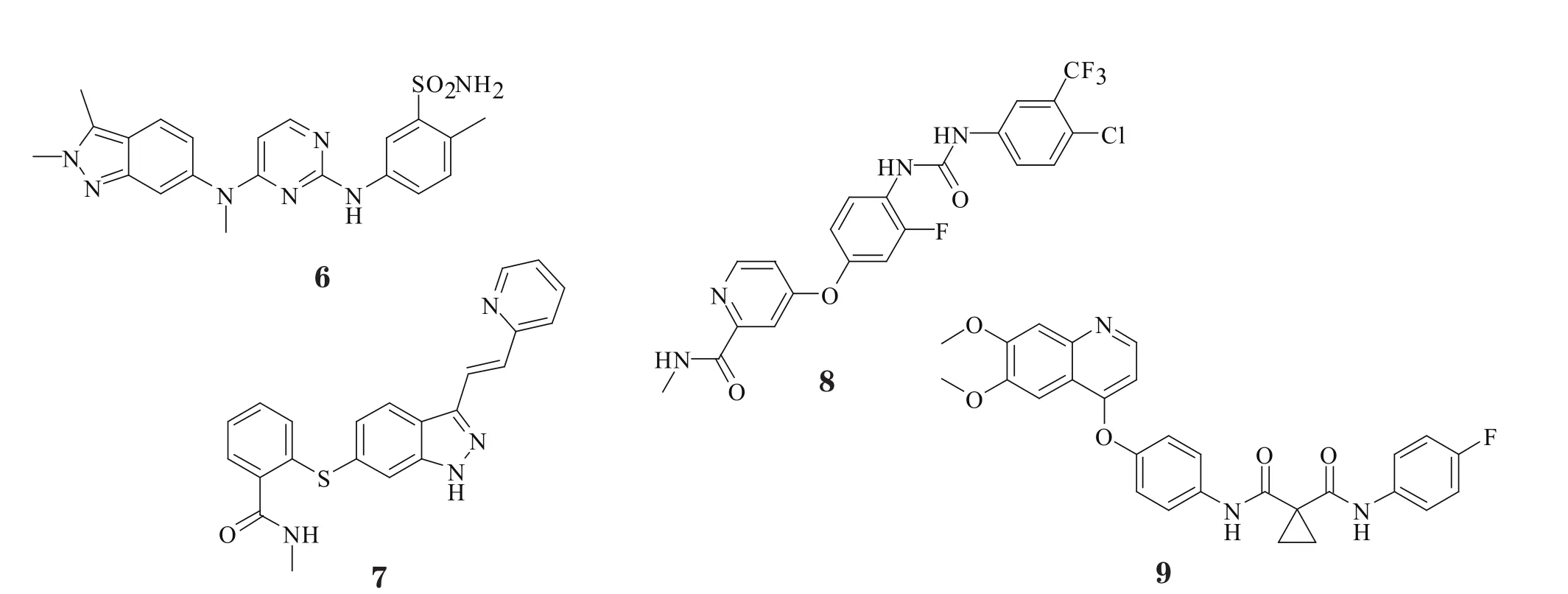
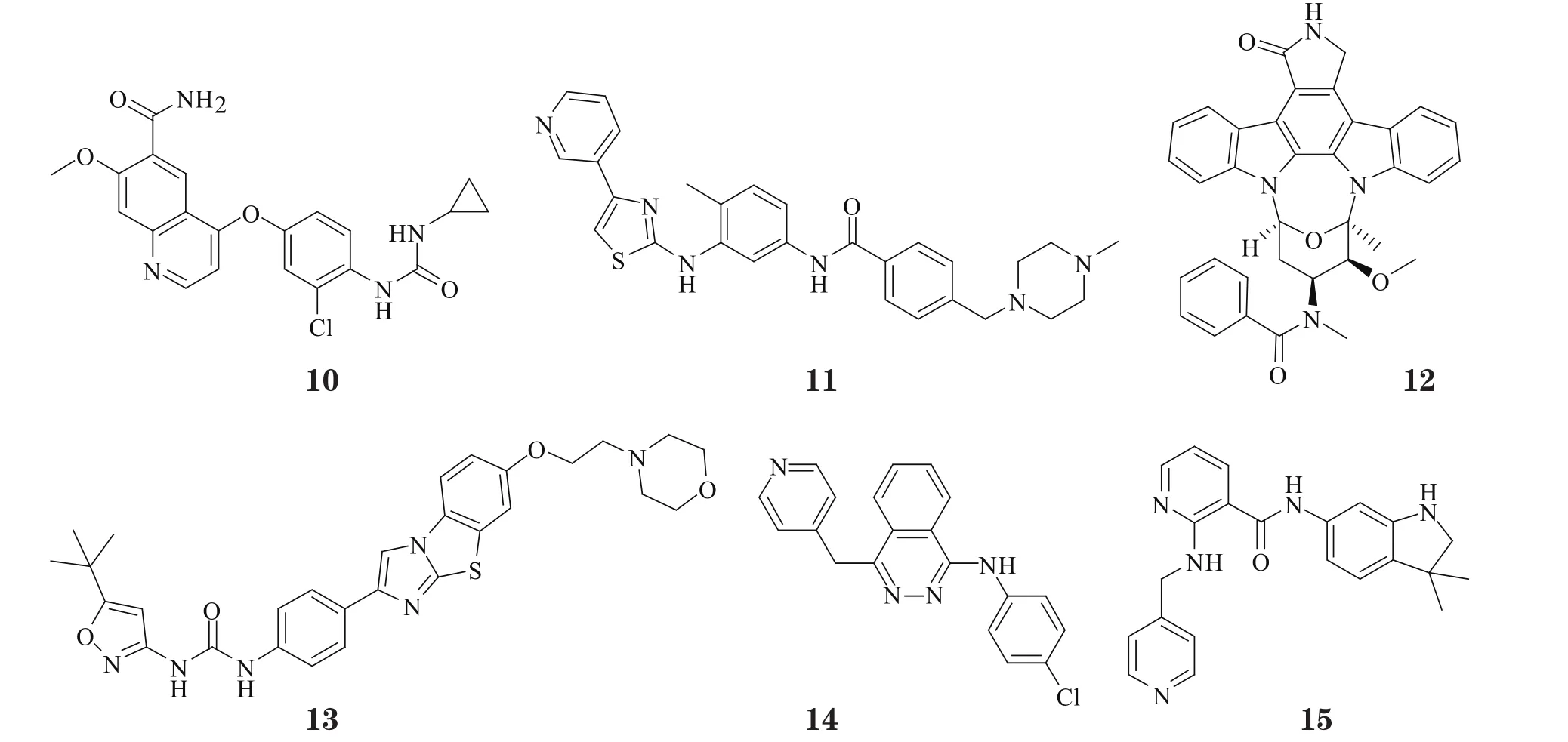
3.2 临床试验阶段的c-Kit 激酶抑制剂目前还有多种不同结构类型的c-Kit 激酶抑制剂正处临床试验阶段(见表2)。
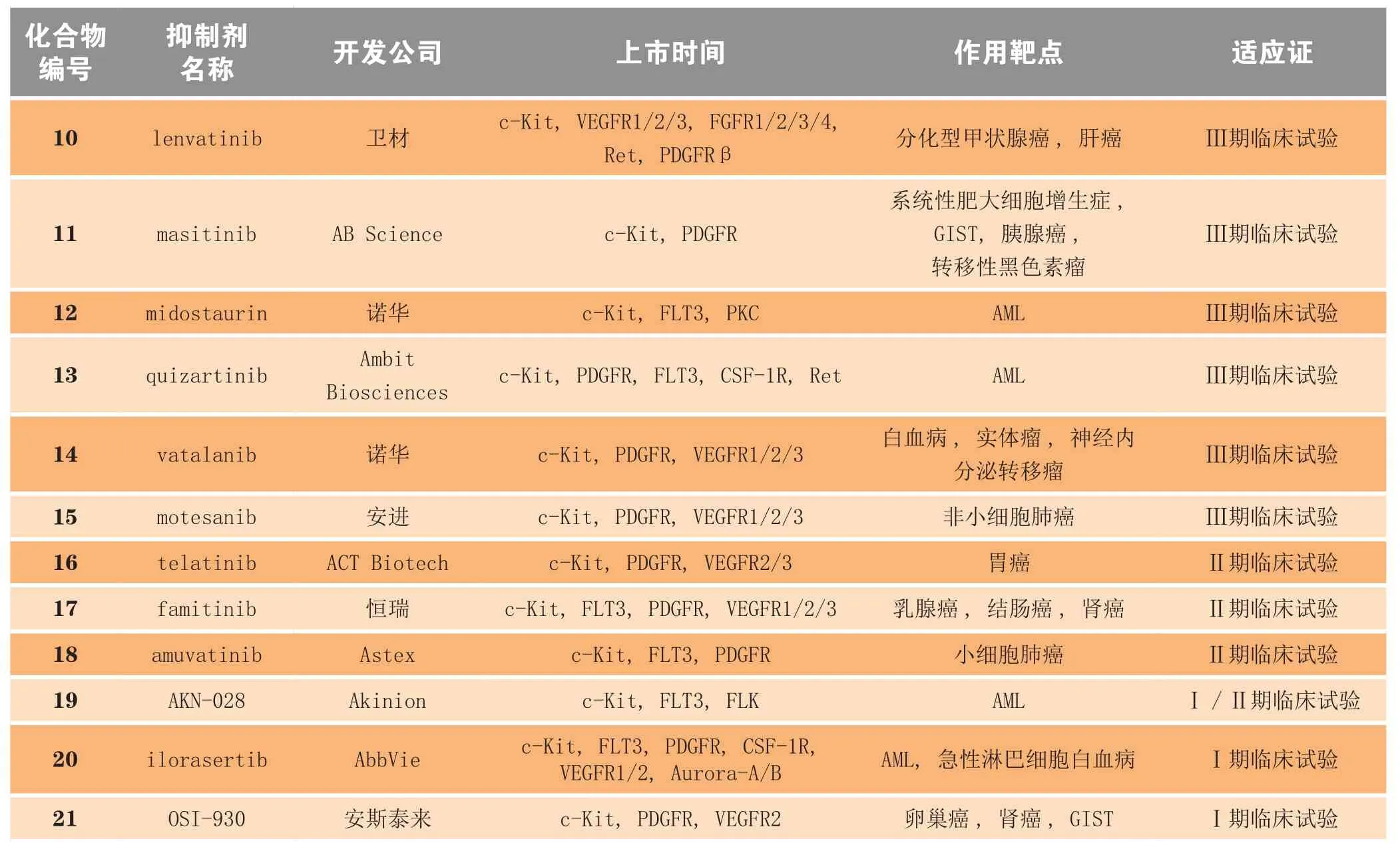
表2 尚处临床试验阶段的c-Kit 激酶抑制剂Table 2 c-Kit kinase inhibitors in clinical trials
Lenvatinib是卫材公司开发的一个多靶点酪氨酸激酶抑制剂,除了与其他抗血管生成药物一样能抑制肿瘤血管生成,还能有效抑制FGFR,FGFR途径的旁路激活被认为是肿瘤细胞逃脱VEGF靶向治疗的机制之一[20]。Ⅰ期临床试验结果显示,lenvatinib的最高耐受剂量为25 mg,口服给药后能快速吸收,3 h左右即达Cmax,并于4周后从体内彻底清除,而其常见的毒副作用为高血压、腹泻、恶心、口腔溃疡、蛋白尿和呕吐[21]。在一项Ⅱ期临床试验中,58位放射性碘难治性分化型甲状腺癌(DTC)患者口服24 mg·d-1lenvatinib,平均治疗周期为405 d,结果显示,有59%的受试者部分应答,36%的受试者病情得到控制,受试者的PFS中值为13.3个月[22]。目前本品正处于一项Ⅲ期临床试验中,该试验在来自欧洲、北美、南美和亚洲等23个国家的392名DTC患者中同时展开[23]。此外,卫材公司还发起了一项全球性的Ⅲ期临床试验,以评估本品用于肝癌的治疗效果[24]。难得的是,日本厚生劳动省、美国FDA和欧洲EMA已先后授予lenvatinib用于治疗甲状腺癌的孤儿药认定资格。
Masitinib是AB Science公司重点开发的一个多靶点酪氨酸激酶抑制剂,能选择性抑制c-Kit和PDGFRα/ β。已结束的一项针对转移性去势抵抗性前列腺癌(mCRPC)的Ⅱ期临床试验显示,masitinib与多西他赛联用能使患者的平均总生存期达到18.4个月,而之前该病患者能达到的最长总生存期仅为13.8个月。为此,AB Science公司在2014年2月宣布开展masitinib与多西他赛联用治疗mCRPC的Ⅲ期临床试验。此外,本品用于治疗其他肿瘤的Ⅲ期临床试验也在进行中[25]。
Midostaurin是诺华公司开发的一种星形孢菌素类酪氨酸激酶抑制剂,不仅能直接抑制肿瘤细胞增殖,还能抑制肿瘤血管的生成。重要的是,本品是目前少有的能有效抑制c-Kit激酶A-loop区D816V突变的抑制剂,且Ⅱ期临床试验证明其对携带c-Kit D816V的SM患者有疗效[26]。目前本品正处于治疗AML的Ⅲ期临床试验阶段。
Quizartinib是一种高效抑制c-Kit、FLT3、PDGFRα/β、Ret和CSF-1R(IC50分别为4.8、1.6、11、7.7、9.9和12 nmol·L-1)的酪氨酸激酶抑制剂。在MV4-11移植瘤小鼠模型实验中,每日1次经口给予本品,连续28 d,结果显示,本品低剂量(1 mg·kg-1·d-1)组模型小鼠的肿瘤生长得到完全抑制,中、高剂量(3、10 mg·kg-1·d-1)组模型小鼠的肿瘤几乎消退,且无一只小鼠出现体质量下降;此外,本品的水溶性和药动学性质也极佳[27]。鉴于2013年结束的一项Ⅱ期临床试验发现复发或难治性的具有FLT3-ITD突变的AML患者对本品可产生高应答率,Ambit Biosciences公司于2014年4月启动了本品的Ⅲ期临床试验[28]。此前,美国FDA和欧洲EMA均已授予本品用于治疗AML的孤儿药认定资格。
Vatalanib是诺华公司开发的血管生成抑制剂,可在纳摩尔级浓度下抑制VEGF诱导的VEGFR2(KDR)自磷酸化以及内皮细胞的增殖、迁移和生存,而在高达 1 μmol·L-1的浓度下对不表达VEGFR的细胞无任何细胞毒性或抗增殖效应。小鼠模型实验显示,经口给予vatalanib(50 mg·kg-1,qd)后,其血浆浓度能在8 h内维持在1 μmol·L-1以上;而在生长因子植入模型以及肿瘤细胞驱动的血管生成模型中,vatalanib(25~100 mg·kg-1,qd,po)对VFGF和PDGF诱导的血管生成呈现剂量依赖性抑制作用,且在相同的剂量范围内,它还能有效抑制裸鼠肿瘤模型中的多种人源性肿瘤的生长以及肿瘤微血管的形成;难得的是,vatalanib对循环血细胞或骨髓粒细胞均无严重损伤作用(Wood等,Cancer Res,2000年)。目前,本品单独使用以及与细胞毒药物联用治疗多种实体瘤的临床试验正在进行,其中包括针对转移性结直肠癌的Ⅲ期临床试验以及针对转移性神经内分泌肿瘤、转移性胃肠道间质瘤、非转移性雄激素非依赖性前列腺癌和弥漫性恶性间皮瘤的Ⅰ期或Ⅱ期临床试验[29]。
Telatinib是 ACT Biotech公 司 重 点 开 发 的VEGFR2/3、c-Kit和PDGFRβ抑制剂。一项telatinib与卡培他滨及顺铂联用治疗晚期胃癌的Ⅱ期临床试验在进行到中期时便出现令人惊喜的结果:2/3可评估的受试者产生迅速而持久的客观肿瘤应答,其中66%的人产生完全应答;且这种联用疗法呈现抗血管生成活性,其耐受性良好,最常见的毒副作用仅表现为轻微或中度疲劳、乏力和胃肠道反应,而只在少于10%的受试者出现严重的高血压、手足综合征和中性粒细胞减少症。基于此,ACT Biotech公司随即启动了telatinib用于一线治疗GIST的Ⅲ期临床试验,该试验设计已得到美国FDA和欧洲EMA的支持,并获得了美国FDA的特别评估协议(SPA)。在此之前,telatinib已被美国FDA授予治疗胃癌的孤儿药认定资格。本品有望于2014年在欧洲和美国上市[30]。
PLX-3397是Plexxikon公司开发的一种选择性作用于c-Kit、FLT3和CSF-1R等Ⅲ型酪氨酸激酶的抑制剂,但其结构尚未公开。本品不仅对FLT-ITD突变体阳性AML有很好的疗效,且对quizartinib耐药的FLT-ITD/ F691L突变体阳性AML仍能保持良好的抑制活性[31]。最新研究显示,在携带c-KitV558del/+突变体的鼠源GIST和人源GIST 移植瘤模型中,PLX-3397不管是在诱导肿瘤细胞凋亡和坏死还是在降低肿瘤体积和质量方面,均明显优于伊马替尼,这可能与其对c-Kit的抑制活性强于伊马替尼有关[32]。在Ⅰ期临床试验中,PLX-3397表现出良好的耐受性,其毒副作用仅为轻微(1级)的贫血、疲劳、恶心和皮疹。目前,本品治疗难治性或复发性霍奇金淋巴瘤的Ⅱ期临床试验已完成,而针对恶性黑色素瘤和转移性乳腺癌等其他肿瘤的Ⅰ/Ⅱ期临床试验正在进行中[33]。
Famitinib是江苏恒瑞公司在舒尼替尼的结构基础上开发出的一种吲哚酮类多靶点酪氨酸激酶抑制剂,能选择性作用于c-Kit、VEGFR2和PDGFRβ(IC50分别为2.3、4.7和6.6 nmol·L-1)。在动物实验中,famitinib显示出广谱抗肿瘤活性,能强效抑制多种移植瘤模型中肿瘤细胞的生长,且与奥沙利铂或5-氟尿嘧啶联用时可显著增强后两者的药效[34]。目前,本品用于治疗肾细胞癌、GIST、胰腺癌和鼻咽癌的Ⅱ期临床试验正在中国展开,且已有的临床研究结果显示,本品能在更低的剂量下达到与舒尼替尼相似的治疗效果[35]。
AKN-028是Akinion公司研发的一种作用于c-Kit、FLT3和FLK的酪氨酸激酶抑制剂。一项针对17种包括骨髓瘤、淋巴瘤、小细胞肺癌、急性淋巴性白血病和AML等不同类型肿瘤细胞的实验显示,AKN-028能选择性抑制所有5种AML细胞株的增殖,其中包括携带c-Kit激活型突变体的Kasumi-1细胞[36]。最近的实验研究显示,AKN-028能下调AML细胞系MV4-11中细胞周期素D依赖性激酶和c-Myc癌基因的表达,将细胞周期阻滞于G0/G1期[37]。本品目前正处于治疗AML的Ⅰ/Ⅱ期临床试验阶段。
4 c-Kit激酶抑制剂耐药机制
c-Kit激酶抑制剂在肿瘤分子靶向治疗中的成功着实令人欢欣鼓舞,然而不幸的是,部分肿瘤患者在使用该类药物一段时间后会出现耐药,其机制十分复杂,主要包括激酶结构域的二次点突变、靶基因的扩增和过度表达或表观遗传学调控激活、冗余/下游信号通路的上调或激活以及ABC转运蛋白的过度表达等[23]。其中,由激酶结构域的二次点突变导致的耐药现象尤为突出,严重影响了现有蛋白激酶抑制剂疗效的发挥。c-Kit耐药型突变主要发生在ATP结合区(外显子13和14)和激酶结构域A-loop区(外显子17)。
4.1 发生在ATP结合区的c-Kit耐药型突变
在用伊马替尼治疗GIST的过程中,约有50%的肿瘤患者出现获得性耐药现象,其主要源于c-Kit耐药型突变[38],其中最常见的是ATP结合区(外显子13和14)的氨基酸点突变,如V654A和T670I,这些突变使得伊马替尼与ATP结合区的亲和力减弱[39]。在c-Kit中,Thr670是ATP口袋铰链区入口的看门残基(gatekeeper),能与伊马替尼形成氢键作用,但当Thr670突变为体积大的异亮氨酸时,这种氢键作用消失,而且突变后产生的空间位阻还会进一步阻碍伊马替尼和其他第2代酪氨酸激酶抑制剂(如尼洛替尼和达沙替尼)与激酶的结合,从而产生耐药现象。另外,c-Kit中的Val654能与伊马替尼形成疏水作用,但当它突变为体积小的丙氨酸后这种疏水作用即消失[39]。为了克服c-Kit中看门残基突变导致的耐药现象,Sugen公司(后被辉瑞公司收购)开发出了多靶点激酶抑制剂舒尼替尼,由于该化合物主要与c-Kit激酶结合位点中的铰链区作用,并不涉及看门残基,不受该位点突变的影响(见图4),故而其能有效抑制具有V654A和T670I突变的c-Kit激酶,因此舒尼替尼被批准用于治疗伊马替尼无效或抵抗的GIST[40]。
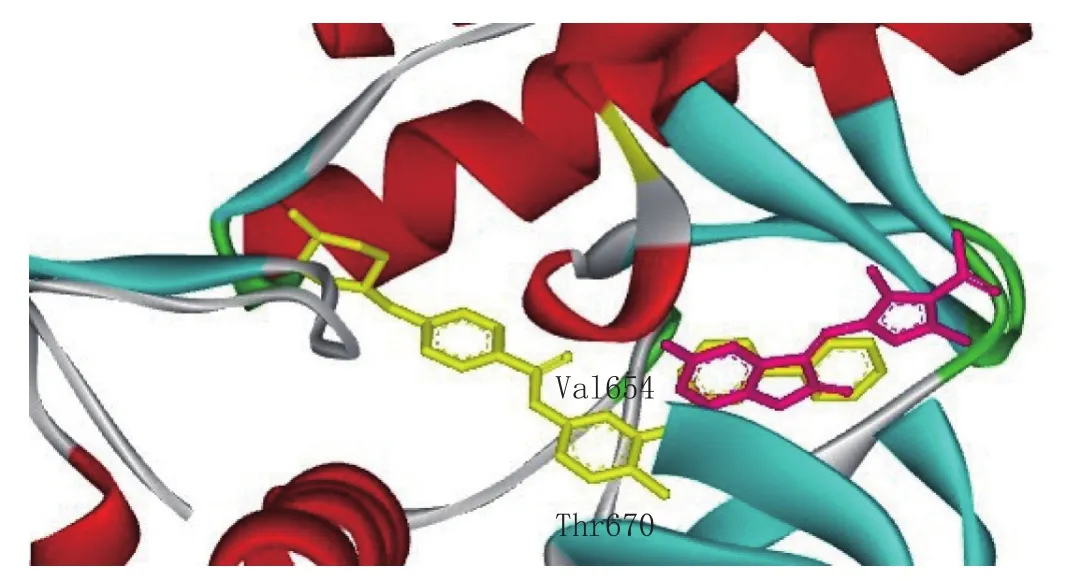
图4 伊马替尼和舒尼替尼与野生型c-Kit激酶的共晶结构叠合图Figure 4 Superimposition of eutectic structures of imatinib and sunitinib bound to wild-type c-Kit kinase
4.2 发生在A-loop区的c-Kit耐药型突变
除了ATP结合区,A-loop区也是c-Kit耐药型突变发生的另一个主要区域,特别是在Asp816、Asp820和Tyr823等残基位点,如D816V突变。外显子17编码的A-loop区对激酶活化构象的调节起十分关键的作用。早期的c-Kit分子建模结果显示,在非活化构象中,负电性的Asp816羧基能稳定带正电荷的一段螺旋偶极子,从而稳定非活化构象,而活化构象则不依赖于这种作用,因此Asp816突变会导致非活化构象中激酶结构域分子间相互作用的不稳定,致使非活化构象转向活化构象[41]。无独有偶,相关动力学研究也证明,突变体c-KitD816H和c-KitD816V的激酶结构域活化速率分别是野生型的184倍和536倍[7]。
上述提到舒尼替尼是具有V654A和T670I突变的c-Kit激酶的强效抑制剂,但后来有研究发现,该药在治疗c-Kit激酶V654A突变体阳性的GIST过程中也会引发c-Kit激酶A-loop区的突变,而此位点突变可导致c-Kit非活化构象的不稳定,促使其向活化构象转变;由于舒尼替尼对激酶活化构象的亲和力非常弱,其只能与激酶的非活化构象结合,因此其诱导的激酶A-loop区突变致使c-Kit对舒尼替尼耐药[42]。
5 结语
多种肿瘤,如GIST、SM、核心结合因子相关性AML和黑色素瘤等的发生发展与c-Kit激活型突变密切相关,因此c-Kit激酶成为一个重要的肿瘤靶向治疗靶标,且c-Kit激酶抑制剂在临床治疗中取得的巨大成功也恰恰说明其极具研究与开发价值。然而,c-Kit激酶抑制剂在肿瘤治疗中的成功并不能掩饰其在应用中出现的获得性耐药问题。因此,深入研究c-Kit激酶抑制剂的获得性耐药机制,开发安全有效的肿瘤治疗策略,乃当务之急。可以相信,随着人们对c-Kit激酶耐药型突变机制的深入探讨和阐明,将有助于促进肿瘤耐药问题的解决,并为新一代c-Kit激酶抑制剂的设计与开发提供坚实的理论基础和新的思路。
[1]Lennartsson J, Rönnstrand L. Stem cell factor receptor/c-Kit: from basic science to clinical implications[J]. Physiol Rev, 2012, 92(4): 1619-1649.
[2]Antonescu C R. The GIST paradigm: lessons for other kinase-driven cancers[J]. J Pathol, 2011, 223(2): 251-261.
[3]Ashman L K, Griffith R. Therapeutic targeting of c-KIT in cancer[J]. Expert Opin Investig Drugs, 2013, 22(1): 103-115.
[4]Opatowsky Y, Lax I, Tomé F, et al. Structure, domain organization, and different conformational states of stem cell factor-induced intact KITdimers[J]. Proc Natl Acad Sci USA, 2014, 111(5): 1772-1777.
[5]Treiber D K, Shah N P. Ins and outs of kinase DFG motifs[J]. Chem Biol, 2013, 20(6): 745 -746.
[6]Lennartsson J, Jelacic T, Linnekin D, et al. Normal and oncogenic forms of the receptor tyrosine kinase kit[J]. Stem Cells, 2005, 23(1): 16-43.
[7]Gajiwala K S, Wu J C, Christensen J, et al. Kit kinase mutants show unique mechanisms of drug resistance to imatinib and sunitinib in gastrointestinal stromal tumor patients[J]. Proc Natl Acad Sci USA, 2009, 106(5): 1542-1547.
[8]Edris B, Willingham S B, Weiskopf K, et al. Anti-KIT monoclonal antibody inhibits imatinib-resistant gastrointestinal stromal tumor growth[J]. Proc Natl Acad Sci USA, 2013, 110(9): 3501-3506.
[9]Miyazawa K. Phosphorylation in the activation loop as the finishing touch in c-Kit activation[J]. J Biochem, 2012, 151(5): 457-459.
[10]Beadling C, Jacobson-Dunlop E, Hodi F S, et al. Kit gene mutations and copy number in melanoma subtypes[J]. Clin Cancer Res, 2008, 14(21): 6821-6828.
[11]Erben P, Schwaab J, Metzgeroth G, et al. The KIT D816V expressed allele burden for diagnosis and disease monitoring of systemic mastocytosis[J]. Ann Hematol, 2014, 93(1): 81-88.
[12]Baccarani M, Deininger M W, Rosti G, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013[J]. Blood, 2013, 122(6): 872-884.
[13]Signorovitch J, Ayyagari R, Reichmann W M, et al. Major molecular response during the frst year of dasatinib, imatinib or nilotinib treatment for newly diagnosed chronic myeloid leukemia: a network metaanalysis[J]. Cancer Treat Rev, 2014, 40(2): 285-292.
[14]Chow P K H, Poon D Y H, Khin M W, et al. Multicenter phase II study of sequential radioembolization-sorafenib therapy for inoperable hepatocellular carcinoma[J]. PloS One, 2014, 9(3): e90909.
[15]Motzer R J, Hutson T E, Cella D, et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma[J]. N Engl J Med, 2013, 369(8): 722-731.
[16]Motzer R J, Escudier B, Tomczak P, et al. Axitinib versus sorafenib as second-line treatment for advanced renal cell carcinoma: overall survival analysis and updated results from a randomised phase 3 trial[J]. Lancet Oncol, 2013, 14(6): 552-562.
[17]Bruix J, Tak W Y, Gasbarrini A, et al. Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: multicentre, open-label, phase II safety study[J]. Eur J Cancer, 2013, 49(16): 3412-3419.
[18]PharmaTimes. Bayer kicks off Ph III Stivarga trial in colorectal cancer patients[EB/OL]. [2014-02-20]. http://www.pharmatimes.com/ Article/14-02-20/Bayer_kicks_off_Ph_III_Stivarga_trial_in_colorectal_ cancer_patients.aspx.http://www.pharmatimes.com/Article/14-02-20/ Baye kicks off Ph III Stivarga trial in colorectal cancer patients.aspx.
[19]Elisei R, Schlumberger M J, Müller S P, et al. Cabozantinib in progressive medullary thyroid cancer[J]. J Clin Oncol, 2013, 31(29): 3639-3646.
[20]Grande E, Diez J J, Zafon C, et al. Thyroid cancer: molecular aspects and new therapeutic strategies[J]. J Thyroid Res, 2012, 2012: 847108.
[21]Boss D S, Glen H, Beijnen J H, et al. A phase I study of E7080, a multitargeted tyrosine kinase inhibitor, in patients with advanced solid tumours[J]. Br J Cancer, 2012, 106(10): 1598-1604.
[22]Sherman S I, Jarzab B, Cabanillas M E, et al. A phase II trial of the multitargeted kinase inhibitor E7080 in advanced radioiodine (RAI)-refractory differentiated thyroid cancer (DTC)[J]. J Clin Oncol, 2011, 29( 15suppl): 5503.
[23]Stjepanovic N, Capdevila J. Multikinase inhibitors in the treatment of thyroid cancer: specific role of lenvatinib[J]. Biologics, 2014, 8: 129-139.
[24]Christina Izzo. Lenvatinib extends PFS in differentiated thyroid cancer[EB/OL]. [2014-02-03]. http://www.onclive.com/web-exclusives/ Lenvatinib-Extends-PFS-in-Differentiated-Thyroid-Cancer#sthash. N08mIOu4.dpuf.
[25]AB Science. AB Science initiation of a new phase 3 study with masitinib in prostate cancer following encouraging survival results obtained in a phase 2 study[EB/OL]. [2014-02-20]. http://globenewswire.com/newsrelease/2014/02/20/612000/10069288/en.
[26]Pardanani A. Systemic mastocytosis in adults: 2011 update on diagnosis, risk stratifcation, and management[J]. Am J Hematol, 2011, 86(4): 362-371.
[27]Chao Q, Sprankle K G, Grotzfeld R M, et al. Identifcation of N-(5-tertbutyl-isoxazol-3-yl)-N′-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b] [1,3]benzothiazol-2-yl]phenyl}urea dihydrochloride (AC220), a uniquely potent, selective, andefficacious FMS-like tyrosine kinase-3 (FLT3) inhibitor [J]. J Med Chem, 2009, 52(23): 7808-7816.
[28]Ambit Biosciences. Ambit announces initiation of quizartinib QUANTUM-R phase 3 trial[EB/OL]. [2014-04-08]. http://www. marketwatch.com/story/ambit-announces-initiation-of-quizartinibquantum-r-phase-3-trial-2014-04-08.
[29]ClinicalTrials.gov.Vatalanib[EB/OL].[2014-05-05].http://www. clinicaltrials.gov/ct2/results?term=vatalanib&Search=Search.
[30]Bicher Cancer Institute. ACT BIOTECH announces positive interim phase 2 results with telatinib in first-line gastric cancer[EB/OL]. [2014-04-08]. http://www.drugs.com/clinical_trials/act-biotechannounces-positive-interim-phase-2-results-telatinib-first-line-gastriccancer-10334.html.
[31]Smith C C, Lin K, Lasater E, et al. Preclinical and clinical resistance mechanisms to the investigational selective FLT3 inhibitor PLX3397 in FLT3-ITD+ acute myeloid leukemia (AML)[J]. Blood, 2013, 122(21): 3938.
[32]Kim T S, Cavnar M J, Cohen N A, et al. Increased KIT inhibition enhances therapeutic effcacy in gastrointestinal stromal tumor[J]. Clin Cancer Res, 2014, 20(9): 2350-2362.
[33]ClinicalTrials.gov.PLX3397[EB/OL].[2014-05-05].http://www. clinicaltrials.gov/ct2/results/displayOpt?flds=a&flds=b&fld s=i&flds=f&flds=c&flds=j&flds=m&flds=p&submit_fld_ opt=on&term=plx3397&show_fds=Y.
[34]Lou L, Mi Y, Xu Y, et al. Preclinical antitumor study of famitinib, an orally available multi-targeted kinase inhibitor of VEGFR/PDGFR/c-Kit in phase I clinical trials[J]. Cancer Res, 2011, 71(8): 3604.
[35]Xie C, Zhou J, Guo Z, et al. Metabolism and bioactivation of famitinib, a novel inhibitor of receptor tyrosine kinase, in cancer patients[J]. Br J Pharmacol, 2013, 168(7): 1687-1706.
[36]Eriksson A, Hermanson M, Wickström M, et al. The novel tyrosine kinase inhibitor AKN-028 has signifcant antileukemic activity in cell lines and primary cultures of acute myeloid leukemia[J]. Blood Cancer J, 2012, 2: e81.
[37]Eriksson A, Kalushkova A, Jarvius M, et al. AKN-028 induces cell cycle arrest, downregulation of Myc associated genes and dose dependent reduction of tyrosine kinase activity in acute myeloid leukemia[J]. Biochem Pharmacol, 2014, 87(2): 284-291.
[38]Demetri G D, Reichardt P, Kang Y K, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial[J]. Lancet, 2013, 381(9863): 295-302.
[39]Tamborini E, Pricl S, Negri T, et al. Functional analyses and molecular modeling of two c-kit mutations responsible for imatinib secondary resistance in gist patients[J]. Oncogene, 2006, 25(45): 6140-6146.
[40]Demetri G D, van Oosterom A T, Garrett C R, et al. Effcacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial[J]. Lancet, 2006, 368(9544): 1329-1338.
[41]Foster R, Griffth R, Ferrao P, et al. Molecular basis of the constitutive activity and sti571 resistance of asp816val mutant kit receptor tyrosine kinase[J]. J Mol Graph Model, 2004, 23(2): 139-152.
[42]Nishida T, Takahashi T, Nishitani A, et al. Sunitinib-resistant gastrointestinal stromal tumors harbor cis-mutations in the activation loop of the kit gene[J]. Int J Clin Oncol, 2009, 14(2): 143-149.
Research and Development of c-Kit Kinase Inhibitors
WANG Peibei, PANG Junxia, LAI Yisheng
(Center of Drug Discovery , China Pharmaceutical University, Nanjing 210009, China)
As a typical member of type III receptor tyrosine kinase family, c-Kit plays a key role in tumor occurrence, development, invasion, migration and recurrence. So it is one of the hot targets for molecular targeted cancer therapy and its inhibitor has become a hot spot of research and development of antitumor drugs. c-Kit kinase and the associations between its activating mutations and tumorigenesis have been introduced. Especially, the recent researches on the c-Kit kinase inhibitors marketed and in clinical trials and their resistance mechanisms have been reviewed.
c-Kit kinase inhibitor; c-Kit activating mutation; c-Kit mutation for drug-resistance; tumor therapy
R730.231; R979.1
A
1001-5094(2014)07-0497-10
接受日期:2014-05-07
*通讯作者: 赖宜生, 教授;
研究方向: 抗肿瘤药物研究;
Tel: 025-83271015; E-mail: yslai@cpu.edu.cn
猜你喜欢
同位素(2022年6期)2022-12-30
物理学报(2022年10期)2022-06-04
人人健康(2021年14期)2021-08-06
唐山师范学院学报(2020年6期)2020-04-16
时代邮刊(2019年22期)2019-12-17
时代邮刊·下半月(2019年11期)2019-09-22
唐山师范学院学报(2019年3期)2019-06-18
中成药(2018年12期)2018-12-29
天然产物研究与开发(2018年1期)2018-02-02
中成药(2018年1期)2018-02-02
