
2013 年全球新药研发报告
——第二部分(Ⅴ)
2014-03-08 06:46GraulNavarroDulsatCrucesTracy
药学进展 2014年7期
Graul A I, Navarro D, Dulsat C, Cruces E, Tracy M
·全球药讯·
GLOBAL PHARMACEUTICAL INFORMATION
2013 年全球新药研发报告
——第二部分(Ⅴ)
Graul A I, Navarro D, Dulsat C, Cruces E, Tracy M

编者按:本刊于2013年分4期译载了汤森路透公司独家授权的“2012年全球新药研发报告”,该报告一经刊出,就因内容全面、资料权威、视角新颖、观点独到、数据翔实、时效性强广受好评。读者纷纷来函索要单行本,众多药企高层对该报告也高度关注。
本期“全球药讯”栏目继续刊登“2013年全球新药研发报告”第二部分(Ⅴ)系列报告和“焦点”中的“聚焦阿尔茨海默病”。相信会为广大读者提供翔实、及时的行业资讯,为启迪研发思路,锁定研发管线助一臂之力!
近年来很多人都悲观地预测医药产业行将消亡,但至今尚未应验,事实上患者活得很好。药品监管者制定的新法规已受到产业的热捧,包括美国食品药品管理局(FDA) 的“突破性疗法”、“合格感染性疾病产品(qualified infectious disease product,QIDP)”认定以及如今在很多国家都已深入人心的孤儿药法规。医药公司往往很务实,及时果断摈弃开发计划中表现欠佳的项目,这样虽对计划进行了瘦身,却提高了项目质量。2013年医药企业仍趋于继续通过并购以巩固在产业中的地位,提升效率。综述2013年医药产业林林总总的发展趋势。
孤儿药;突破性疗法认定;合格感染性疾病产品;生物仿制药;中止开发;撤市
改变一个成熟产业的方向,堪比驾驭“泰坦尼克号”。不可否认,这是一个缓慢而艰辛的过程,药品监管机构与医药公司的共同努力正开始显现成果。如今绕过了冰山,整个产业并未触礁沉没,前些年的种种悲观预言并未应验。相反,2013 年却成为 10 多年来首次上市新药和生物制品数量最多的一年(见图1),而近 10 年里来出现类似佳绩的年份也只有 2009 年,当时正值H1N1流感泛滥,刺激了新型流感疫苗的快速开发和批准,其数量创下历史记录。2013 年则是新型流感疫苗红红火火的另一年份,有5 种流感新疫苗上市[1],但其所占当年上市新药品总数的份额小于2009年的表现(5/56 vs 11/50,即 9% vs 22%)。
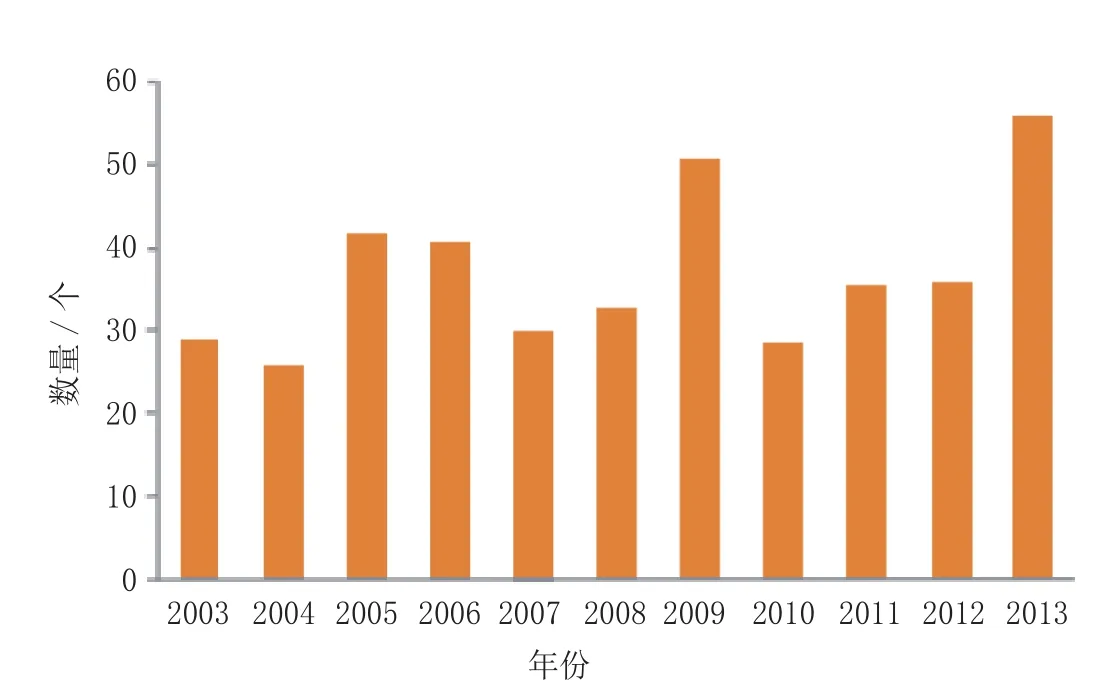
图1 2003—2013年首次上市的新药和生物制品总数量比较Figure 1 Comparison of the total numbers of new drugs and biologics launched for the first time during 2003-2013
2013 年,医药产业除了在新品上市数量方面取得了令人瞩目的成绩,还收获了其他若干振奋人心的开发新成果,表明该产业不仅保持着良好的生存活力,而且正开始适应变幻莫测的全球环境。
美国FDA与制药公司及患者群体合作,积极主动实施新法规,加快对用于严重和危及生命以及目前尚无法治疗疾病的新药的开发和批准。在几十年前对孤儿药立法的基础上,美国FDA近来再次兑现了在这一领域的承诺,创立了2个新法规,以刺激被忽视领域的药物开发,即突破性疗法认定和QIDP认定,现概述如下。
1 孤儿药
孤儿药即罕见病用药,当属世界首例的《美国孤儿药法案》签署生效已过去30 多年,如今该法案已显见成效。孤儿药与传统意义上的重磅药品是相对立的,且已不再是一个新生事物。眼下,对罕见病治疗药物的研究正在医药产业掀起热潮,不仅只有小公司主攻或专攻孤儿药的开发,跨国大公司也在慢慢转型,纷纷通过转让和(或)收购方式投入巨资。于是,目前罕见病治疗药物的开发已在较短时间内呈指数增长,2013年首次上市的新药中有 20 个品种在上市国拥有“孤儿药”资格(见表1) ,这是近年来所见的最大数量,如图2 所示。
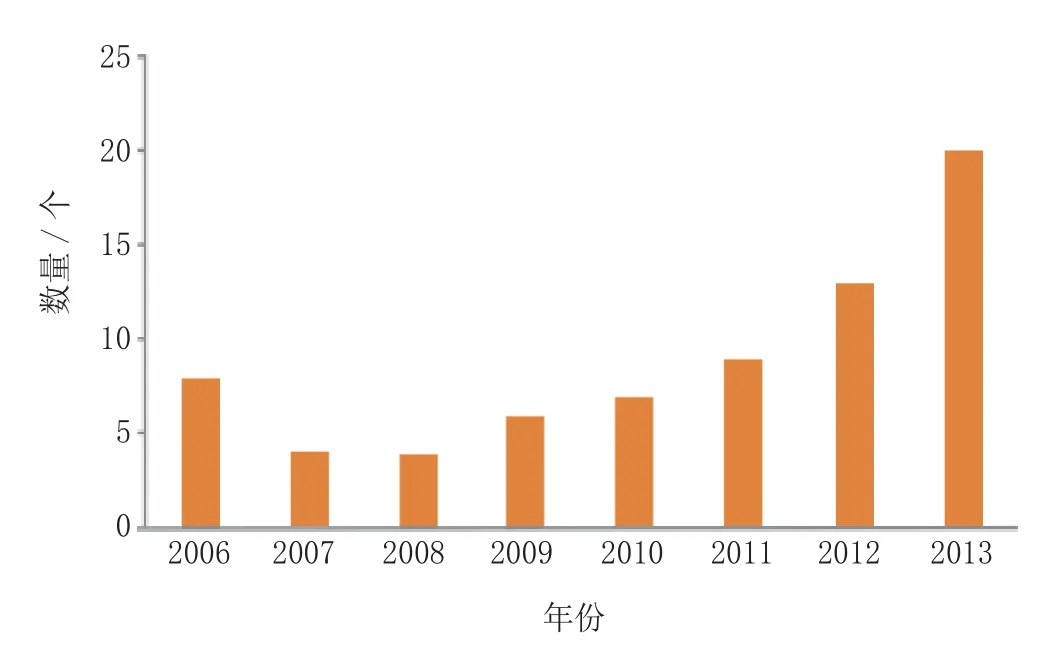
图2 2006—2013年首次上市的新孤儿药数量比较Figure 2 Comparison of the numbers of new orphan drugs launched for the first time during 2006-2013

表1 2013年首次上市的新孤儿药Table 1 New orphan drugs launched for the first time in 2013
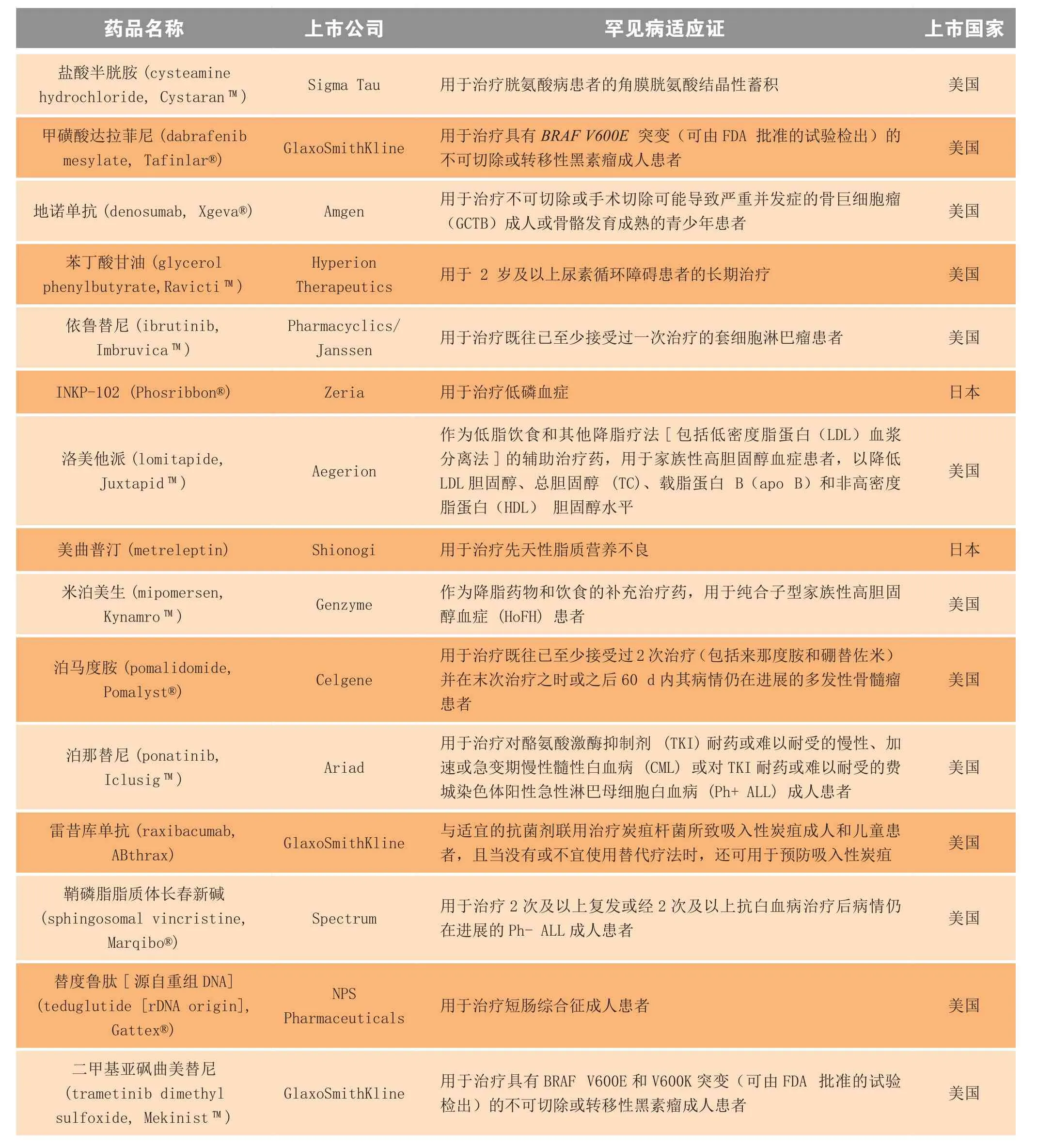
续表1
新孤儿药认定的颁布节奏让人目眩,仅在2013 年,实行孤儿药法规的国家(如欧美、日本和澳大利亚等)药品监管机构就颁发了225 个以上的新孤儿药认定,其中部分摘选见表2。
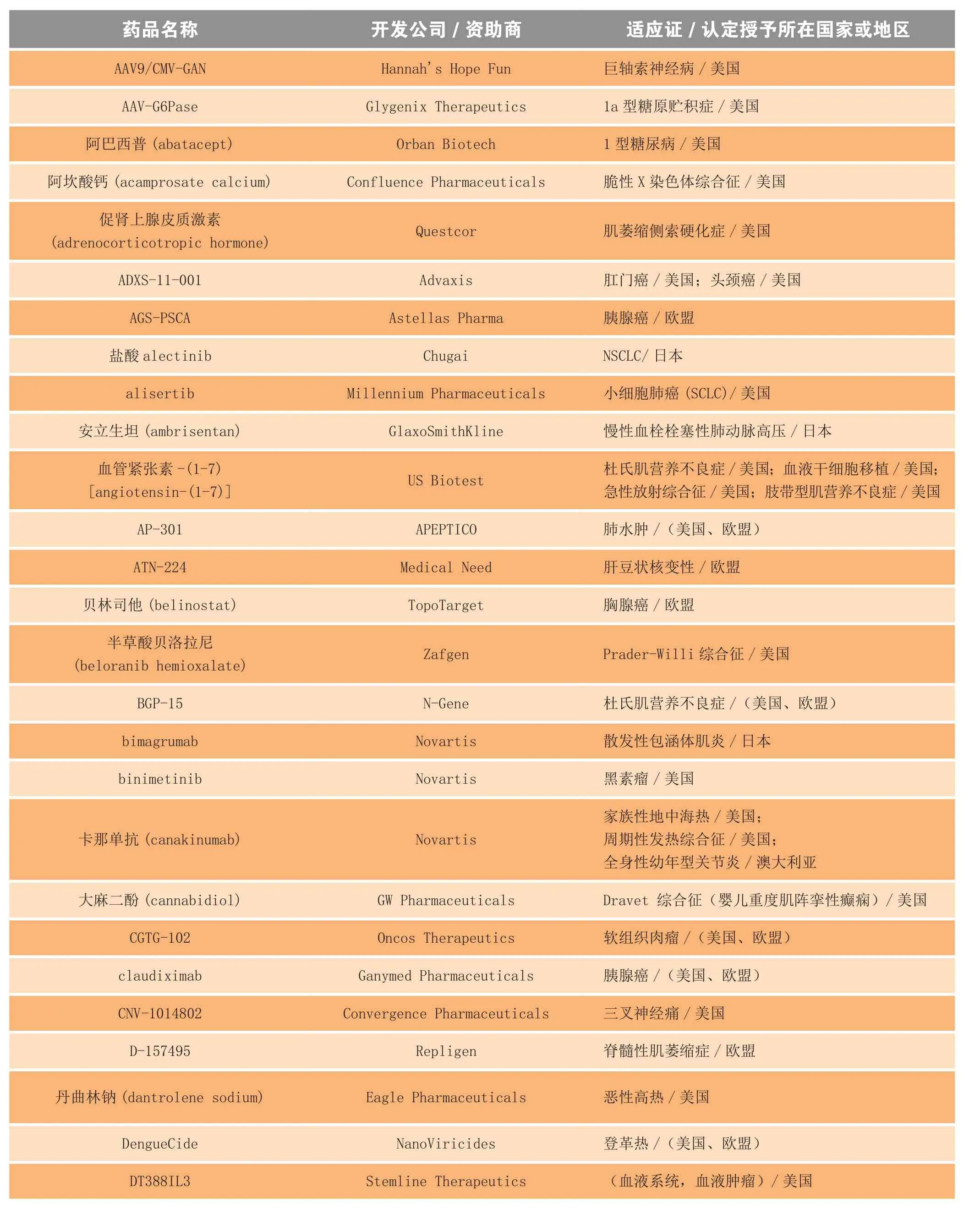
表2 2013年获得孤儿药认定的部分药物和生物制品Table 2 Selected drugs and biologics receiving orphan drug designation in 2013
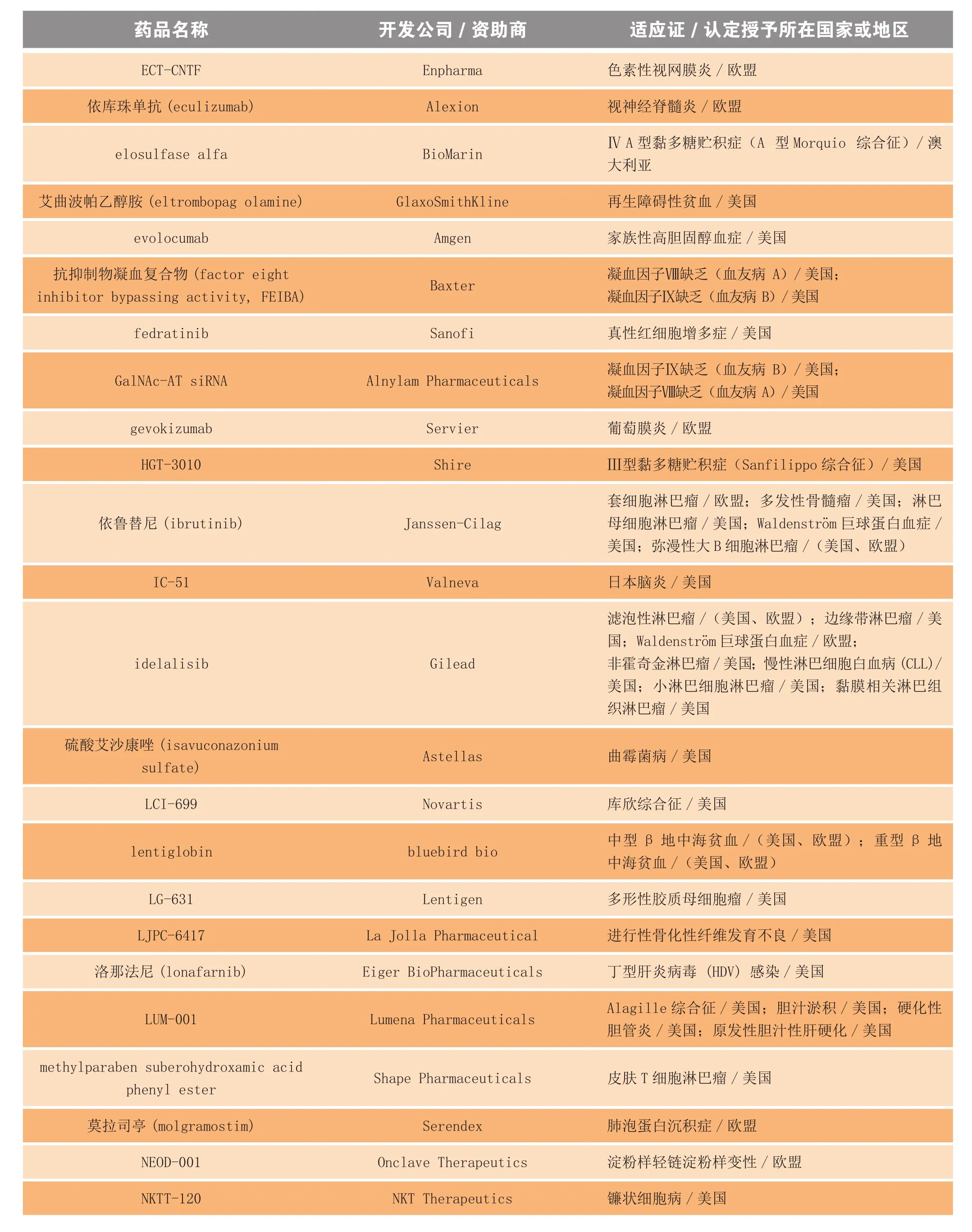
续表2
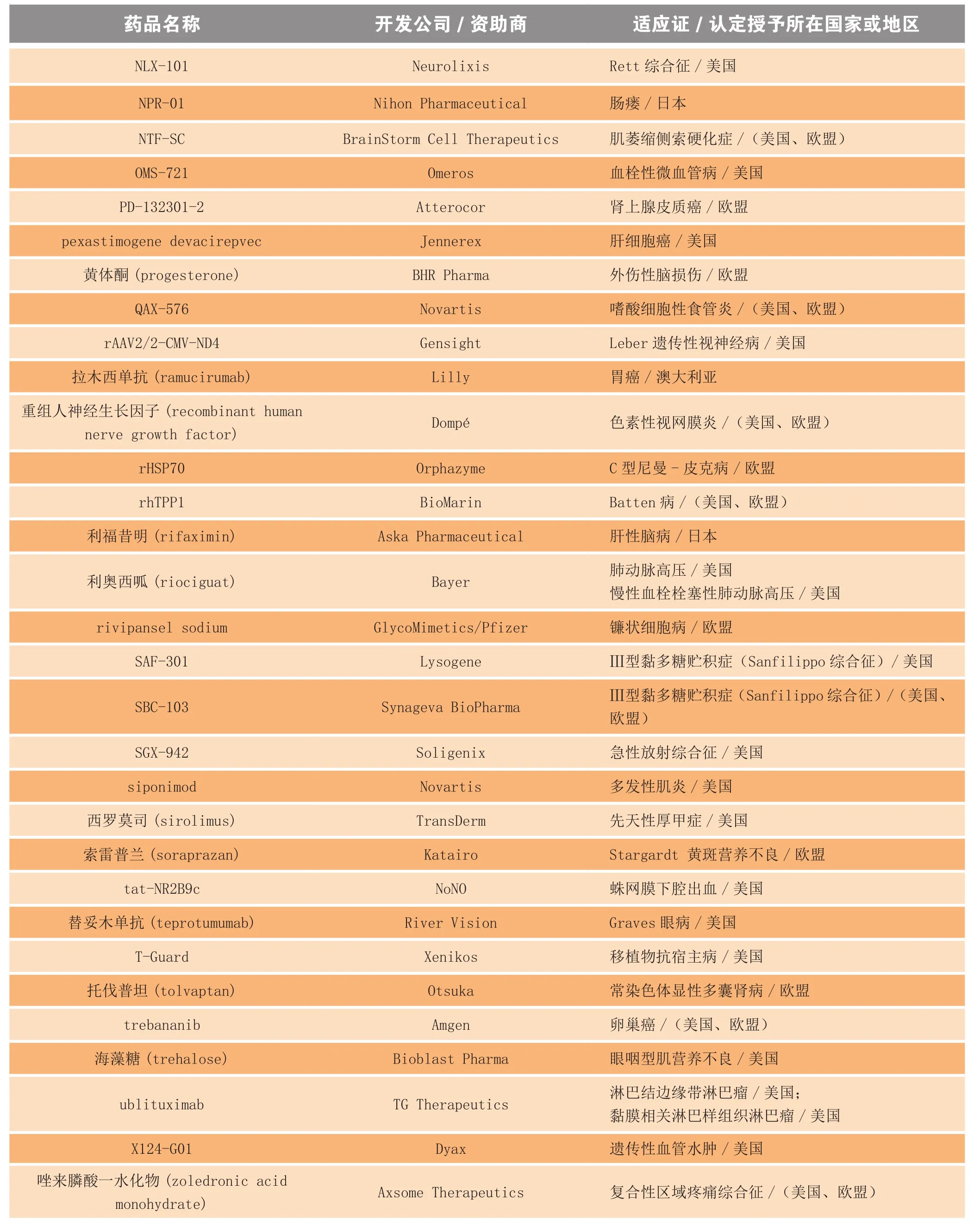
续表2
制药公司将注意力转向孤儿药的主要原因之一,是意识到其盈利能力不断增加。罕见病患者即便有治疗选择,也屈指可数,因此只有为孤儿药支付高价。例如,2013年在美国推出的短肠综合征治疗药替度鲁肽(teduglutide,Gattex®)其1年的治疗费用是29.5万美元。 更令人咋舌的是,2005 年上市的加硫酶(galsulfase,Naglazyme)其1年的治疗费用高达44.1万美元,目前仍是Ⅵ型黏多糖增多症患者的唯一治疗药[2]。各家公司纷纷辩称,这些药品高价是有道理的,原因是与糖尿病和哮喘等更为常见的疾病患者相比,这些药品的受众面小得多。
现已知需要治疗的罕见病有7 000多种,孤儿药开发持续增长的空间巨大,但仅凭盈利因素可能并不足以维持这种驱动力。比利时研究人员在2014年1月发表的一篇文章指出,现今孤儿药厂商的业绩并不强于传统制药公司,实际盈利还不如后者。尽管孤儿药厂商的平均毛利润比非孤儿药厂商高11%,但其还得拿出很大一部分销售利润再次投入到新的研发,结果拉低了其经营获利能力及投资回报率[3]。
孤儿药开发中遇到的另一个难题是临床试验的设计和实施。由于可参与临床试验评价新孤儿药的患者人数一般很少,因此要招募到能令人信服地证明孤儿药疗效的足够多受试者,可能会成为一大艰巨任务。再者,多数罕见病都是严重或危及生命的,故在临床研究中使用安慰剂对照会引发伦理道德问题,而替代终点的使用会令试验结果的解释和推断更为困难。此外,急于将新药推向市场的意愿,会促使临床试验周期缩短,以致无法考察到生存期改善等长期疗效指标。在全球主要药品市场,对孤儿药开发的关注度与日俱增,上述这些关键问题必须尽早解决[4]。
2 突破性疗法认定
美国FDA 新近推出的突破性疗法认定是一系列举措中最新出台的法规,以加快严重疾病治疗药物的开发和评审,其比早先出台的法规(即快速通道认定、加速批准及优先评审)更进了一步,据称特别适用于促进分子靶向治疗药的开发[5]。
该法规作为《美国FDA安全和创新法案 (FDASIA)》的一部分于 2012 年 7 月签署生效[6]。依据2013年6月发布的指南草案,“若某药被开发单用或与一种或多种其他药物联用于治疗严重或危及生命的疾病,且初步临床试验表明,在一个或多个有临床意义的终点指标上,该药较现有疗法有显著改善,如临床开发早期观察到的明显疗效。”那么该药开发公司或资助者便能申请突破性疗法认定,而被确认具“突破性疗法认定”资格的药品也就能享受一系列优惠待遇,包括快速通道认定的所有特权、FDA官员的悉心指导(早在Ⅰ期临床开发阶段便可开始,以推进一项高效药物开发计划的实施)以及有高级管理者和资深评审人员参与对开发计划进行积极协作性的跨学科评审[7]。指南最终版本有望于 2014 年夏季出炉。
此法规提出,对于临床开发早期便呈现显著活性的治疗药物予以加速评审,并鼓励开发商在不晚于Ⅱ期临床开发阶段就去申请突破性疗法认定。但若药物在开发后期未能达到早期期望值,FDA则可撤销其候选药资格。2013 年1月,Vertex 公司的囊性纤维化治疗药ivacaftor 和lumacaftor成为首批接受突破性疗法认定的药物,而截至年底,有3个药物作为首批拥有该认定资格的药物获批上市:阿托珠单抗(obinutuzumab,Roche/Genentech公司)、依鲁替尼(ibrutinib,Johnson & Johnson/Pharmacyclics 公司)和索非布韦(sofosbuvir,Gilead 公司)。
根据与药物开发公司的信息交流而收录的获突破性疗法认定的部分药品名单见表3,美国FDA 目前尚未正式发布突破性候选药物的官方名单。
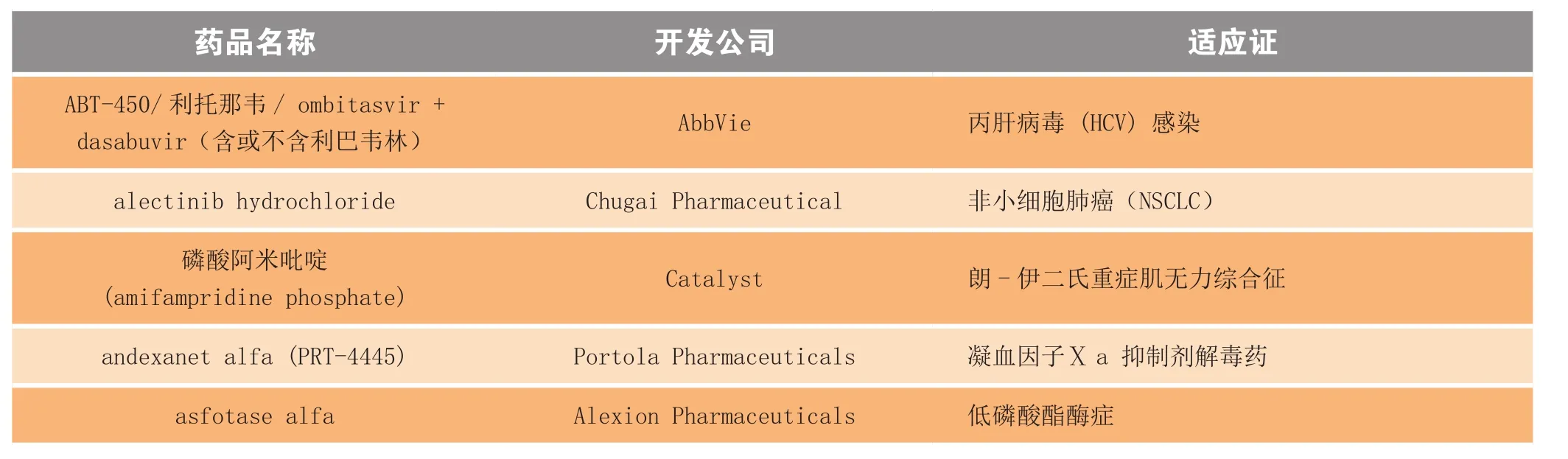
表3 2013年获得突破性疗法认定的部分药品Table 3 Partial drugs receiving breakthrough designation in 2013
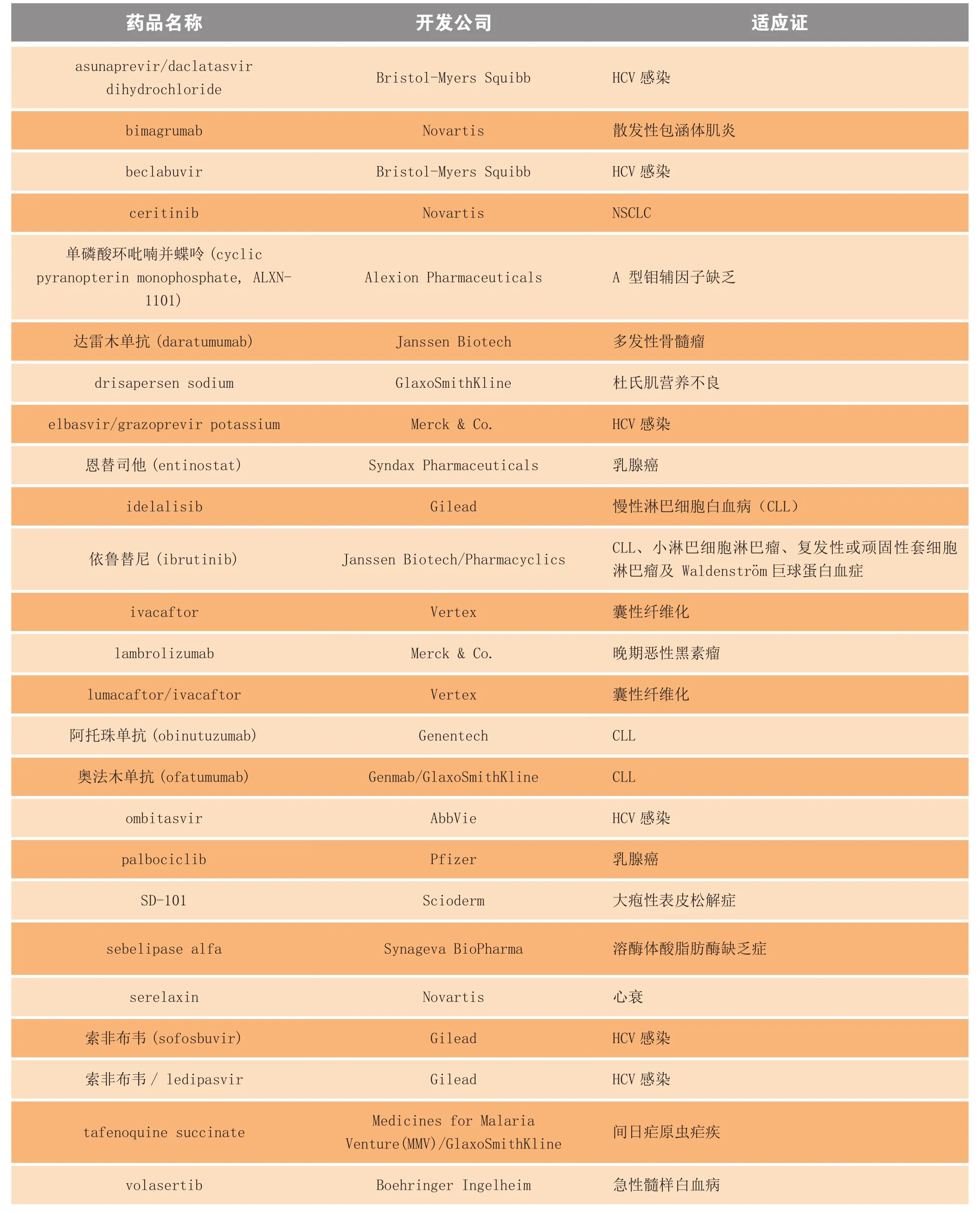
续表3
突破性疗法认定可授予针对同一适应证的多个在研药品,但当用于此适应证的首个药物获批后,用于同一适应证的任何其他药物都将失去突破性疗法资格,除非药物开发公司或资助者能证明其产品优于首个获批药品[8]。可以想象,在不远的将来,这一状况会很快出现,因为如表3所示,目前有多个候选药品的适应证是丙型肝炎和慢性淋巴细胞白血病 (CLL) 等。
3 合格感染性疾病药品
同样,按 FDASIA 第Ⅷ篇章创立的《抗生素激励法案(Generating Antibiotics Incentives Now,GAIN)》旨在刺激新型抗生素的开发,以解决不断增长的感染性疾病威胁。对于那些开发用于治疗包括由抗菌或抗真菌药耐药病原菌(包括新出现的病原菌)引起的严重或危及生命的感染的抗菌和抗真菌药物,除了享有以前《联邦食品药品化妆品法》赋予的市场独占期外,该新法规还赋予其5 年市场独占期,而且这些药品还拥有快速通道和优先评审资格,它们即被认定为QIDP。
2013 年 6 月,美国FDA 首席科学家 Jesse L. Goodman博士在非洲、全球卫生、全球人权及国际组织美国小组委员会前的证言中称,截至当月,FDA 已给 17 个药品授予了 QIDP 资格,其中相应的 12 种不同活性组分也享受GAIN赋予的待遇[9]。
表4列出2013 年取得QIDP资格的部分药物,此一览表根据与药物开发公司的信息交流而汇编。
4 生物仿制药
生物仿制药亦称“生物相似药”或“后续生物药”,是与原研生物药相似的药物,但绝不会完全相同。化学仿制药可在实验室里进行分析,以证实其确为化学药复制品,而生物仿制药则不然。与传统化学药相比,生物药分子更大,结构复杂得多,大多由活体细胞产生,因此使用当前的科学方法,既不能获得现有生物药的完全复制品,亦不能证明两者之间的可互换性。
目前,全球10多个国家已有第1代生物药的生物仿制药上市,主要是在欧盟,欧洲药品管理局(EMA)自2003年以来就已建立了适宜的生物仿制药审批体系。日本厚生劳动省于2009年批准了其首个生物仿制药。然而,在美国,尚无此类产品注册,也尚无正式途径支持对较为复杂的生物药的仿制品(美国称“后续生物药”)的审批,但现已有至少16份研究性新药申请(IND)被批准给予申报的对应于11种原创品牌生物药的生物仿制药[10]。不过,美国对生物仿制药的监管现状在不远的将来可望有所改变,原因是2012年最高法院批准了《患者保护与平价医疗法案 (PPACA)》。PPACA 第Ⅷ篇章包含了《生物药价格竞争和创新法案(BPCIA)》,其构建了对后续生物药的简化审批程序的监管框架,与对化学仿制药相似。该审批程序要求,开发商或资助者需证明申报的后续生物药与原创参照品之间在功能和结构上的相似及差异之处[11]。
在所有生物制品中,单克隆抗体 (MAb) 的开发尤为复杂。在欧盟,生物仿制MAb的开发受制于特殊指南,其要求有更为严格的临床试验及免疫原性评价[12]。2013年欧盟委员会批准了首批生物仿制MAb:Celltrion 公司的Remsima™和Hospira公司的Inflectra™,两者均为抗肿瘤坏死因子-α(TNF-α)MAb英利昔单抗(infliximab,Remicade®)的生物仿制产品。
经济刺激是开发生物仿制药的主要驱动力量,从原创品牌生物药的销售数据便可见一斑(见表5)。由这份数据可见,迄今最受青睐的生物药是TNF-α拮抗剂依那西普 (etanercept),其原创品牌产品(Enbrel®;Amgen/Pfizer公司)在 2012 年的销售额为 85亿美元。Cipla 公司于2013年在印度上市了一款依那西普生物仿制药,其适应证涵盖了Enbrel®所获准的全套适应证。除此以外,还有其他几款针对一个或多个适应证的依那西普生物仿制药正在开发中(见表6)。
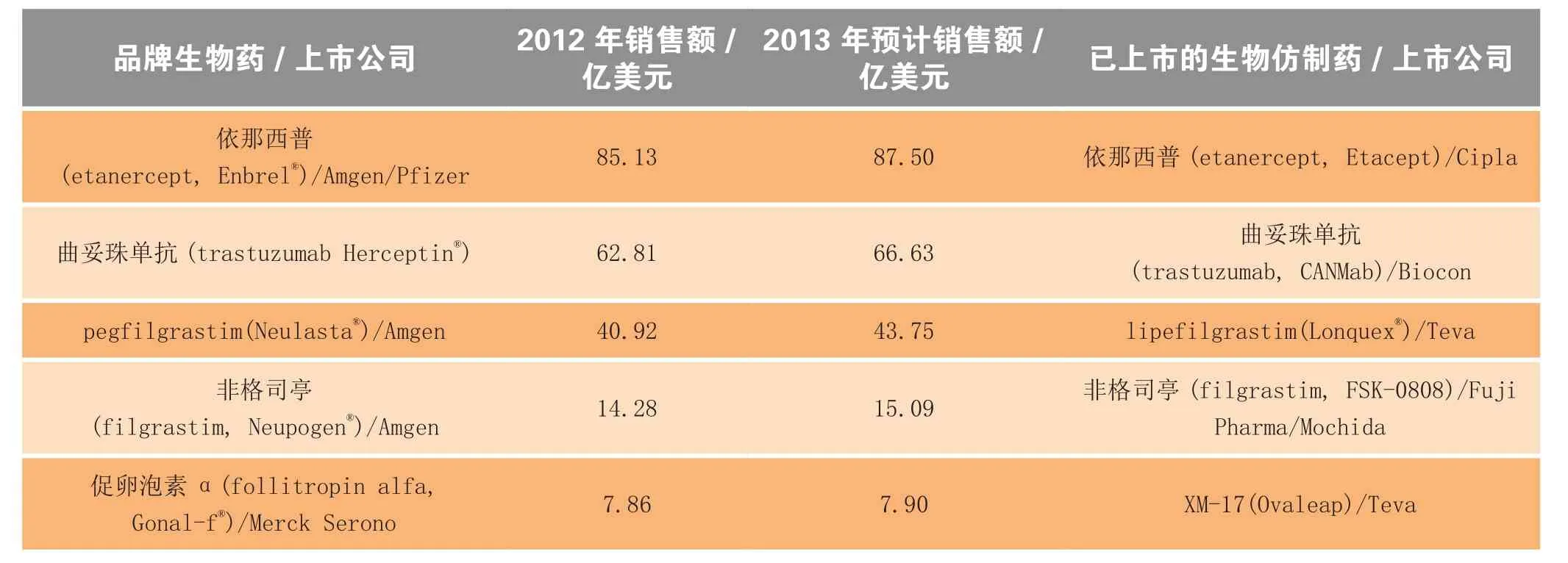
表5 面临生物仿制药新竞争的原创品牌生物药近2年市场销售额Table 5 Sales of brand biologics facing new competition from biosimilars in recent two years
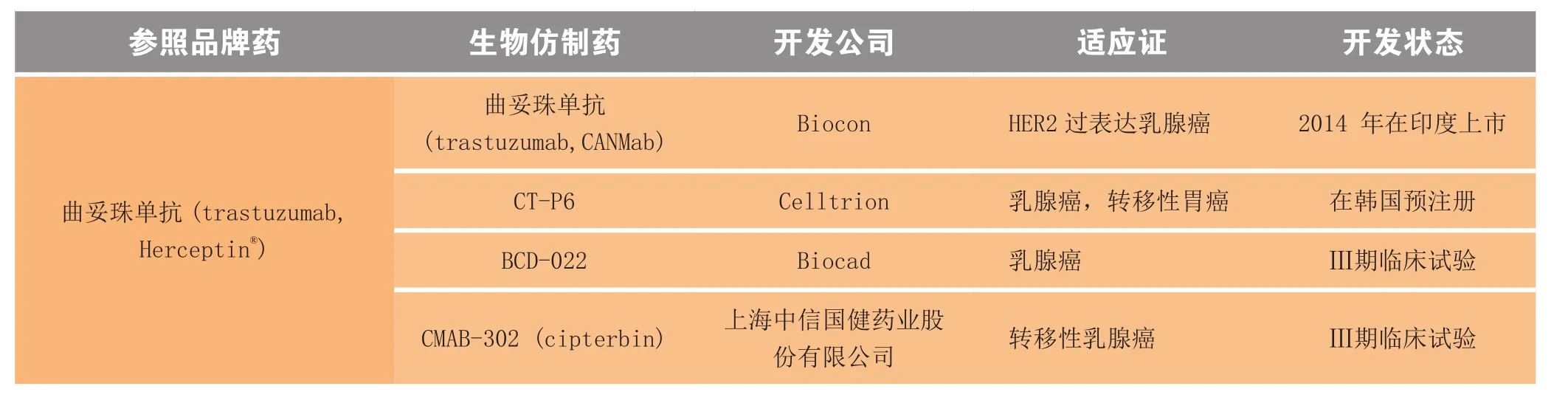
表6 2013年获批上市及正处后期临床试验阶段的部分生物仿制药Table 6 Selected biosimilars and follow-on biologics approved for marketing in 2013 and in late-stage clinical trial
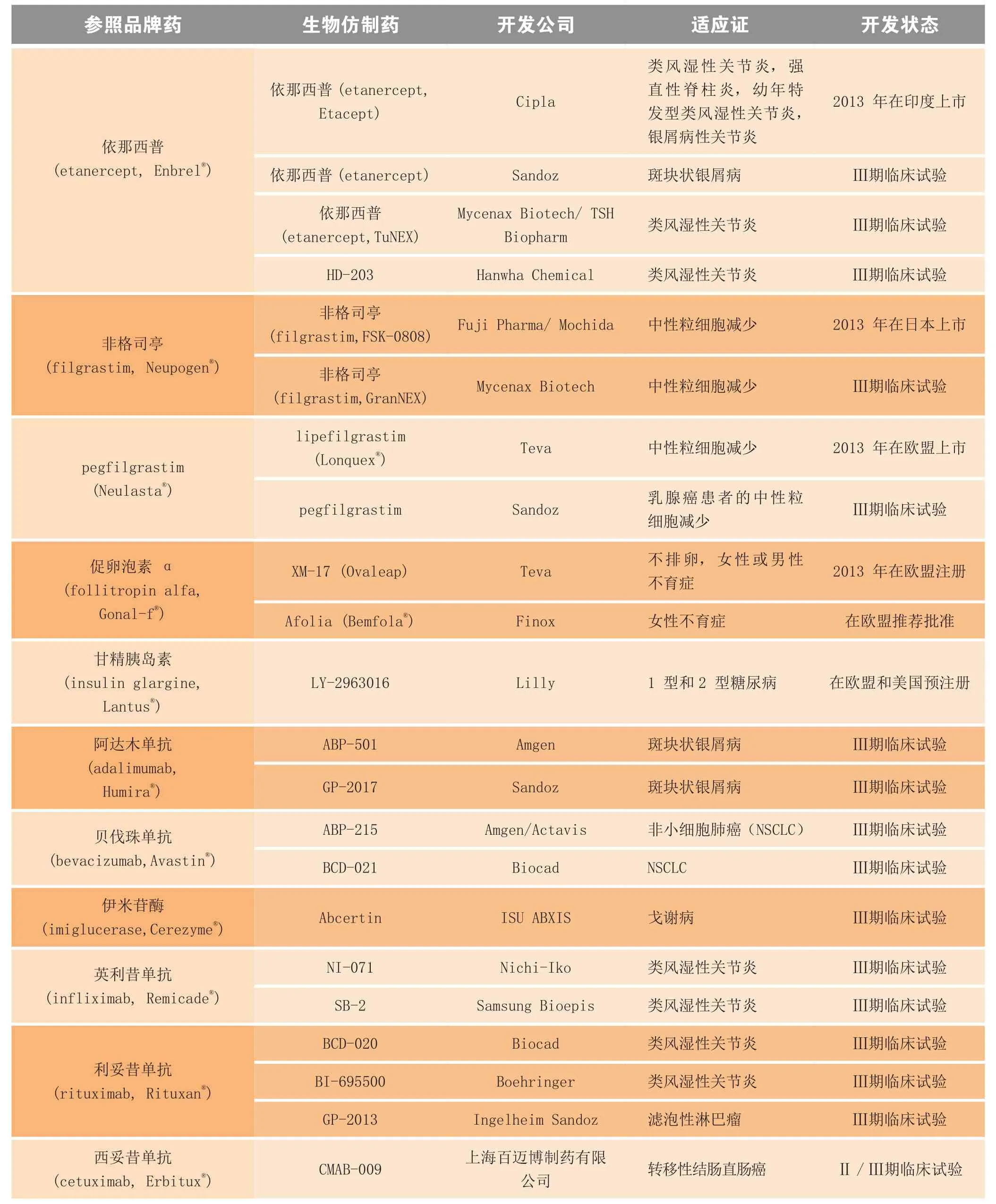
续表6
尽管传统仿制药制造商迫不及待地想进入生物仿制药这一新市场,但与传统药物相比,生物药在很多方面都不易遭受仿制药的竞争。首先,生物药的制造工艺复杂,即使对于原创此工艺的公司亦是如此,生物仿制药生产厂商更是永远无法精准重复原创生物药的制造工艺,因为这属于专有信息;除了复杂的制造工艺外,以生物仿制药谋利受限的其他因素还包括原创生物药的市场独占期漫长、其原创公司面对仿制药竞争时可能采取降价措施以及生物仿制药与其原创参照药品之间缺乏可互换性而致使仿制药可能无法成为替代品且其适应证也难以推定。在生物药市场,仿制药参与竞争所面临的一个主要障碍就是高昂的开发费用,因此与小分子仿制药相比,其投资回报率可能较低。
生物仿制药有可能明显节省医疗费用,这是其获得政府支持的一个重要因素,但各个国家对不同生物仿制药产品的理解和使用会有所不同。例如,在欧盟,生物仿制药虽仅占可进入市场11%的份额,但有些国家却在较为广泛地使用这些药品。据国际遗传流行病学学会进行的一项研究报道,从2007年到2020年间,仅欧洲8国(德国、法国、英国、瑞典、意大利、西班牙、波兰和罗马尼亚)对生物仿制药使用的增长就将累计节省118亿到334亿美元,而主要亚洲市场对生物仿制药的引入也正在不断增长[10]。
表6概括了2013 年处于后期临床试验以及上市和批准的生物仿制药产品。
A Report of New Drugs Research and Development in 2013——Part II(V)
Graul A I, Navarro D, Dulsat C, Cruces E, Tracy M
The demise of the pharmaceutical industry, so pessimistically predicted by many people in recent years, has not come to pass and in fact the patient is alive and well.New programs enacted by drug regulators have been enthusiastically taken up by the industry, including the FDA’s breakthrough therapy and qualifed infectious disease product(QIDP) designations,as well as the now-consolidated orphan drug programs in many countries.Pharma companies pragmatically wean nonperformers from the pipeline in an effcient manner,resulting in somewhat leaner but higher-quality pipelines.Mergers and acquisitions also continue to drive consolidation and effciency in the industry,a trend that continued during 2013.This article provides an updated review of these and other trends in the pharmaceutical industry in the year just passed.
orphan drug; breakthrough therapy designation; qualifed infectious disease product; biosimilar; discontinuation; withdrawal
R97 [关键词]A
1001-5094(2014)07-0527-12
猜你喜欢
包装工程(2022年18期)2022-09-27
今日农业(2022年13期)2022-09-15
当代经济(2016年26期)2016-06-15
转化医学电子杂志(2015年4期)2015-12-27
小说月刊(2015年11期)2015-04-23
创业家(2015年9期)2015-02-27
创业家(2015年9期)2015-02-27
中国卫生(2014年8期)2014-11-12
中国火炬(2012年12期)2012-07-24
中国医药生物技术(2012年5期)2012-02-03
