
Why Zinc?A Density Functional Reactivity Theory Study on Metal-Binding Specificity of Zinc Finger Proteins
2013-12-22 05:21:22ZHAODongboRONGChunyingLIANShixunLIUShubin
湖南师范大学自然科学学报 2013年2期
ZHAO Dong-bo,RONG Chun-ying*,LIAN Shi-xun,LIU Shu-bin,2*
(1.Key Laboratory of Chemical Biology and Traditional Chinese Medicine Research(Ministry of Education of China)and Key Laboratory of Resource Fine-Processing and Advanced Materials of Hunan Province,College of Chemistry and Chemical Engineering,Hunan Normal University,Changsha 410081,China 2.Research Computing Center,University of North Carolina,Chapel Hill,North Carolina 27599-3420,USA)
The zinc finger,a ubiquitous protein-nucleic acid recognition motif invariantly conserved in eukaryote proteins,is a globular minidomain containing a tetrahedral metal-binding site coordinated by cysteine and histidine residues.Its geometry has been well studied by EXAFS[1],spectrophotometrics,NMR[2-4],and synthetic models[5-6].Experimental evidence has unequivocally showed that specific nucleic acid binding activities of these proteins depend on the availability of zinc ions[7-10].Removal of zinc with chelating agents could result in a complete loss of the specific DNA binding activity,while the addition of Zn2+,but not other similar divalent first-row transition metal ions such as Mn2+,Fe2+,Co2+,Ni2+,and Cu2+,would restore the reactivity.The reason behind is unknown.In this work,density functional reactivity theory(DFRT)reactivity indices,which are conceptually insightful and practically convenient in predicting chemical reactivity and regioselectivity of a molecule,are applied to elucidate the metal-binding specificity of zinc fingers.
Previous work[11-15]has shown that DFRT is capable of elucidating many chemical phenomena and predicting molecular acidity and basicity.In DFRT[16-19],chemical potential μ and hardness η are defined as the first and second-order partial derivatives of the total energy E with respect to the total number of electrons N and with the external potential ν(r)fixed,respectively,μ=-χ=(∂E/∂N)νand η=(∂2E/∂N2)ν.Chemical potential μ is the negative of the electronegativity(χ)[20],and softness S is defined as the reciprocal of hardness,S=1/η.According to Mulliken[21],one has μ=-χ=-(1/2)(I+A)and η=I-A[22],where I and A are the first(vertical)ionization potential(IP)and electron affinity(EA),respectively.Under the Koopman's theorem for the closedshell molecules,based on the finite difference approach,I and A can be approximated by the highest occupied molecular orbital(HOMO)energy εHOMO,and the lowest unoccupied molecular orbital energy εLUMO,respectively,I≈-εHOMO;A≈-εLUMO.Recently,Parr,Szentpály,and Liu[23]introduced the concept of electrophilicity index,ω,in terms of μ and η,ω=μ2/2η,appraising the capacity of an electrophile to accept the maximal number of electrons in a neighboring reservoir of electron sea.More recently,Ayers and co-workers[24-25]have proposed two new reactivity indices to quantify nucleophilic and electrophilic capabilities of a leaving group,nucleofugality ΔEn=-A+ω=(μ+η)2/2η and electrofugality ΔEe=I+ω=(μ-η)2/2η.
The models of the zinc-finger proteins studied in this work,abbreviated as MS4,MS3N1,and MS2N2(where M=Mg,Ca,Sc,Ti,V,Cr,Mn,Fe,Co,Ni,Cu,Zn;S and N stand for His and Cys residues,respectively)were taken from the RCSB Protein Data Bank[26],with the PDB IDs 1NJ3,2LO3,and 1WO4,respectively.These three models are shown in Scheme 1.
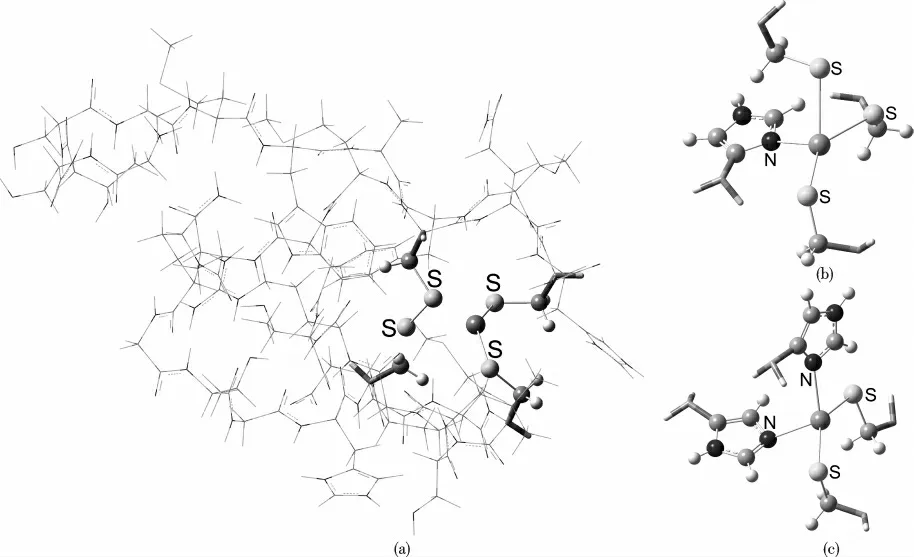
Scheme 1 A three-layer ONIOM model for MS4 in subunit(a)and two truncated high layer models for MS3N and MS2N2 in subunits(b)and(c),respectively with a divalent transition metal ion in the center of each motif.Visualizations of the molecular structures were rendered using GaussView 5.0.Color code:S,yellow;N,blue;C,gray;H,white.
The ONIOM(Our own N-layered Integrated molecular Orbital and molecular Mechanics)model was employed to make the calculations tractable for the geometry optimization with each of the systems in a higher spin state,followed bya harmonic vibrational frequency analysis to confirm that the structures obtained were indeed a minimum on the potential energy surface.The semiempirical PM6 approach[27]was used for the middle layer;the molecular mechanics UFF(universal force field)method[28]was employed for the low layer,and the high layer,treated at the DFT B3LYP/6-31G(d)level of theory[29-31],consists of the divalent metal ion and the ligand atoms(S and N)from the His and Cys residues.All quantum chemical calculations both for structures and properties were performed with the GAUSSIAN-09 package[32]with tight self-consistent field(SCF)convergence criteria,ultrafine integration grids and without symmetry constraints.
Tables 1-3 summarize the results of DFRT indices for these three zinc-finger protein motif models,MS4,MS3N1,and MS2N2.In Tab.1,εHOMOenergy and electronegativity χ are negative in values,while other quantities,such as the lowest molecular orbital energy εLUMO,chemical potential μ,hardness η,softness S,electrophilicity ω,electrofugality ΔEe,and nucleofugality ΔEnare all positive in values.One of the key thermodynamic parameters that explains why our Nature favors zinc rather than other metal ions is hardness,which is the largest in Tab.1 among all the species studied in this work.It is known that the larger the hardness the more stable the system[33],indicating that the zinc-finger motif with the zinc cation binded to it possesses the best stability.This is the first side of this zinc-finger-motif coin,stability.Now,let us look at the other side of the coin,reactivity.As shown by the electronegativity χ,electrophilicty ω,electrofugality ΔEe,and nucleofugality ΔEnindices in Tab.1,the species containing the Zn ion shows again the largest value in each of these quantities,suggesting that the zinc-finger motif with zinc cation in place exhibits the most reactivity in these categories of molecular reactivity.Put together,these results from the two sides of the coin show that when the zinc finger has the zinc ion in place,it possesses the most stability and at the meanwhile most reactivity.This seamless combination of often-contradictory properties of stability and reactivity at the same time in the same place is the unique feature of the zinc-finger motif.
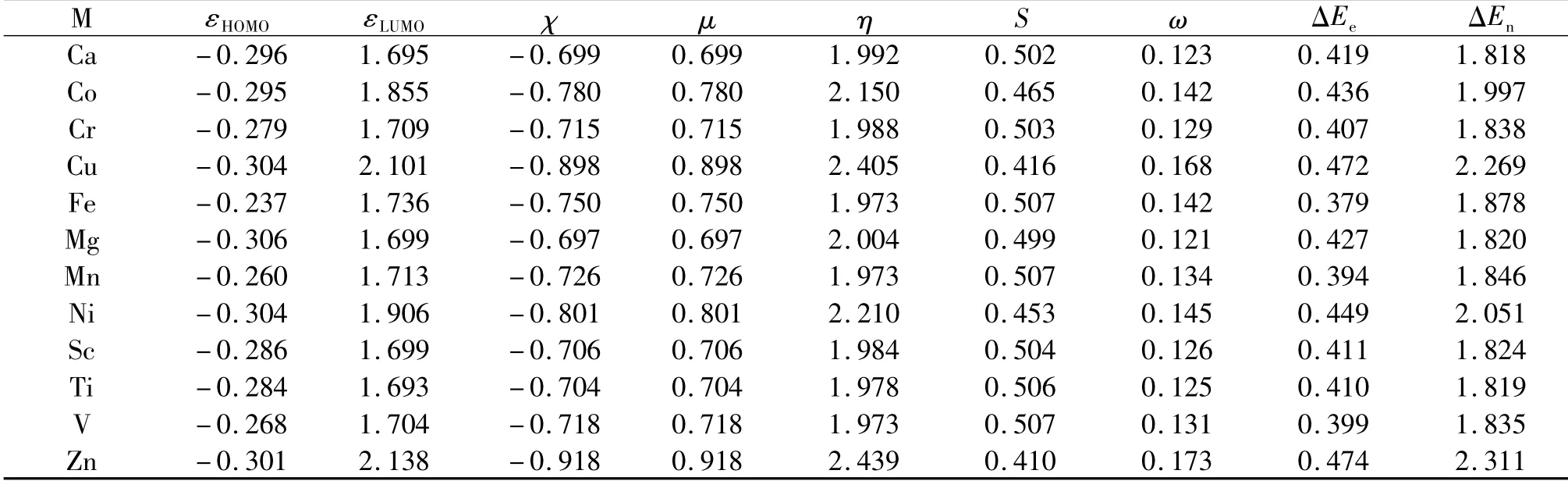
Tab.1 Shown here are for 12 divalent metal ions their highest occupied molecular orbital(HOMO)energy,lowest unoccupied molecular orbital(LUMO)energy,electronegativity(χ),chemical potential(μ),hardness(η),softness(S),electrophilicity(ω),electrofugality(ΔEe),and nucleofugality(ΔEn)indices for the zinc-finger protein motif MS4.Units in atomic units
Next,let us take a look of another zinc finger protein motif model,MS3N1,whose DFRT results are shown in Tab.2.Are the trend and conclusion still the same?The answer is yes.In this case,hardness is still the largest for the zinc ion and electronegativity χ,electrophilicty ω,electrofugality ΔEe,and nucleofugality ΔEnindices are still the largest or second largest in values as well.These same trends confirm that for the second category of the zinc finger motif,both the most stability and best reactivity still remarkably coexist in the same system at the same time.
Finally,we switch our focus to the third zinc-finger motif model,MS2N2,as shown in Tab.3.As can be seen from the Table,the same trend and same conclusion is still valid for systems from this model,where hardness is still the largest for the species with the zinc ion in place whereas its reactivity indices such as electronegativity χ,electrophilicty ω,electrofugality ΔEe,and nucleofugality ΔEnare the largest or second largest.Again,these results verify the conclusion we drew earlier that the uniqueness of the zinc-finger motif is its spectacular combination of two contradictory properties of a molecular system,stability and reactivity.

Tab.2 Shown here are 12 divalent metal ions,highest occupied molecular orbital(HOMO)energy,lowest unoccupied molecular orbital(LUMO)energy,electronegativity(χ),chemical potential(μ),hardness(η),softness(S),electrophilicity(ω),electrofugality(ΔEe),and nucleofugality(ΔEn)for the zinc-finger protein motif MS3N1.Units in atomic units

Tab.3 Shown here are 12 divalent metal ions,highest occupied molecular orbital(HOMO)energy,lowest unoccupied molecular orbital(LUMO)energy,electronegativity(χ),chemical potential(μ),hardness(η),softness(S),electrophilicity(ω),electrofugality(ΔEe),and nucleofugality(ΔEn)for the zinc-finger protein motif MS2N2.Units in atomic units
In summary,our present work employing density functional reactivity theory indices unambiguously shows that the unique feature of the zinc-finger motif is its seamless combination of stability and reactivity.This remarkable property of zinc-singer motifs explains nicely the metal-binding specificity of the zinc-finger proteins.As to how the reactivity is impacted and why this combination is essential,more studies are in need and still in progress,whose results will be published elsewhere.
[1]DIAKUN G P,FAIRALL L,KLUG A.EXAFS study of the zinc-binding sites in the protein transcription factor ⅢA[J].Nature,1986,324(6098):698-699.
[2]FRANKEL A D,BERG J M,PABO C O.Metal-dependent folding of a single zinc finger from transcription factor ⅢA[J].Proc Natl Acad Sci USA,1987,84(14):4841-4845.
[3]PÁRRAGA G,HORVATH S J,EISEN A,et al.Zinc-dependent structure of a single-finger domain of yeast ADR1[J].Science,1988,241(4872):1489-1492.
[4]HARPER L V,AMANN B T,VINSON V K,et al.NMR studies of a cobalt-substituted zinc finger peptide[J].J Am Chem Soc,1993,115(7):2577-2580.
[5]CORWIN D T JR,FIKAR R,KOCH S A.Four-and five-coordinate cobalt(II)thiolate complexes:models for the catalytic site of alcohol dehydrogenase[J].Inorg Chem,1987,26(19):3079-3080.
[6]CORWIN D T JR,GRUFF E S,KOCH S A.Zinc,cobalt,and cadmium thiolate complexes:models for the zinc(S-cys)2(his)2centre in transcription factor ⅢA(cys=cysteine;his=histidine)[J].J Chem Soc Chem Commun,1987(13):966-967.
[7]HANAS J S,HARUDA D J,BOGENHAGEN D F,et al.Xenopus transcription factor A requires zinc for binding to the 5 S RNA gene[J].J Biol Chem,1983,258(23):14120-14125.
[8]KADONAGA J T,CARNER K R,MASIARZ F R,et al.Isolation of cDNA encoding transcription factor Sp1 and functional analysis of the DNA binding domain[J].Cell,1987,51(6):1079-1090.
[9]NAGAI K,NAKASEKO Y,NASMYTH K,et al.Zinc-finger motifs expressed in E.coli and folded in vitro direct specific binding to DNA[J].Nature,1988,332(6161):284-286.
[10]EISEN A,TAYLOR W E,BLUMBERG H,et al.The yeast regulatory protein ADR1 binds in a zinc-dependent manner to the upstream activating sequence of ADH2[J].Mol Cell Biol,1988,8(10):4552-4556.
[11]FENG X T,YU J G,LIU R Z,et al.Why iron?A spin-polarized conceptual density functional theory study on metal-binding specificity of porphyrin[J].J Phys Chem A,2010,114(21):6342-6349.
[12]LIU S B,ESS D H,SCHAUER C K.Density functional reactivity theory characterizes charge separation propensity in protoncoupled electron transfer reactions[J].J Phys Chem A,2011,115(18):4738-4742.
[13]KUMAR N,LIU S B,KOZLOWSKI P M.Charge separation propensity of the coenzyme B12-tyrosine complex in adenosylcobalamin-dependent methylmalonyl-CoA mutase enzyme[J].J Phys Chem Lett,2012,3(8):1035-1038.
[14]HUANG Y,ZHONG A G,LIU S B.Predicting pKavalues for singly and multiply substituted benzoic acids with density functional reactivity theory[J].J Nat Sci Hunan Normal Univ,2011,34(1):52-55.
[15]HUANG Y,LIU L H,LIU S B.Towards understanding proton affinity and gas-phase basicity with density functional reactivity theory[J].Chem Phys Lett,2012,527(0):73-78.
[16]PARR R G,YANG W.Density-functional theory of atoms and molecules[M].New York:Oxford University Press,1989.
[17]GEERLINGS P,DE PROFT F,LANGENAEKER W.Conceptual density functional theory[J].Chem Rev,2003,103(5):1793-1874.
[18]CHATTARAJ P K,SARKAR U,ROY D R.Electrophilicity index[J].Chem Rev,2006,106(6):2065-2091.
[19]LIU S B.Conceptual density functional theory and some recent developments[J].Acta Phys Chim Sin,2009,25(3):590-600.
[20]PARR R G,DONNELLY R A,LEVY M,et al.Electronegativity—the density functional viewpoint[J].J Chem Phys,1978,68(8):3801-3807.
[21]MULLIKEN R S.A new electroaffinity scale;together with data on valence states and on valence ionization potentials and electron affinities[J].J Chem Phys,1934,2(11):782.
[22]PARR R G,PEARSON R G.Absolute hardness:companion parameter to absolute electronegativity[J].J Am Chem Soc,1983,105(26):7512-7516.
[23]PARR R G,SZENTPÁLY L V,LIU S B.Electrophilicity index[J].J Am Chem Soc,1999,121(9):1922-1924.
[24]AYERS P W,ANDERSON J S M,RODRIGUEZ J I,et al.Indices for predicting the quality of leaving groups[J].Phys Chem Chem Phys,2005,7(9):1918-1925.
[25]AYERS P W,ANDERSON J S M,BARTOLOTTI L J.Perturbative perspectives on the chemical reaction prediction problem[J].Int J Quantum Chem,2005,101(5):520-534.
[26]RCSB Protein Data Bank[DB/OL].[2012-12-10].http://www.pdb.org/pdb/home/home.do.
[27]STEWART J J P.Optimization of parameters for semiempirical methods.V.Modification of NDDO approximations and application to 70 elements[J].J Mol Model,2007,13(12):1173-1213.
[28]RAPPÉ A K,CASEWIT C J,COLWELL K S,et al.UFF,a full periodic-table force-field for molecular mechanics and molecular-dynamics simulations[J].J Am Chem Soc,1992,114(25):10024-10035.
[29]BECKE A D.Density-functional exchange-energy approximation with correct asymptotic-behavior[J].Phys Rev A,1988,38(6):3098-3100.
[30]BECKE A D.Density-functional thermochemistry.Ⅲ.The role of exact exchange[J].J Chem Phys,1993,98(7):5648-5652.
[31]LEE C,YANG W,PARR R G.Development of the Colle-Salvetti correlation energy formula into a functional of the electron density[J].Phys Rev B,1988,37(2):785-789.
[32]FRISCH M J,TRUCKS G W,SCHLEGEL H B,et al.Gaussian 09,Revison B.01;Gaussian Inc.:Wallingford,CT,2009.
[33](a)CHATTARAJ P K,LEE H,PARR R G.HSAB principle[J].J Am Chem Soc,1991,113(5):1855-1856;(b)PARR R G,CHATTARAJ P K.Principle of maximum hardness[J].J Am Chem Soc,1991,113(5):1854-1855.
