
BO2团簇超卤素特性的实验和理论研究
2013-12-19 13:18许洪光候高垒孔祥玉
中国科学技术大学学报 2013年5期
冯 源,许洪光,候高垒,孔祥玉
(1.合肥师范学院电子信息工程学院,安徽合肥 230601;2.中国科学院化学研究所分子反应动力学国家重点实验室,北京 100190)
BO2团簇超卤素特性的实验和理论研究
冯 源1,2,许洪光2,候高垒2,孔祥玉2
(1.合肥师范学院电子信息工程学院,安徽合肥 230601;2.中国科学院化学研究所分子反应动力学国家重点实验室,北京 100190)
超卤素团簇特殊的稳定性和物理化学性质使得它特别适合作为制造新型团簇组装型材料的基元.BO2团簇的电子亲和能达到了4.46 eV,同时它的中性团簇差一个电子达到满壳层,具备了成为超卤素的条件.采用光电子能谱与密度泛函理论计算相结合的方法,研究了BO2与Na,Cu原子的相互作用,以及所形成的NaBO2团簇的水溶性.结果表明,在BO2与Na,Cu原子相互作用所形成的CuBO2和NaBO2团簇中,BO2仍然保持直线型结构并且表现出与卤素相似的性质,所形成团簇的电子特性也与卤盐类似.另一方面,NaBO2团簇的水溶性也与卤盐的溶解特性一致,刚开始以紧密离子对(CIP)形式存在,在结合了3个水分子以后,光电子能谱有了很明显的改变,对应着Na+与(OBO)-的溶剂隔离的离子对结构(SSIP)的出现.并且,在实验过程中,还出现了电子亲和能更大的Cu(BO2)2团簇(5.07 eV),为了与超卤素相区别,我们定义其为长链二级超卤素,该团簇由3种元素组成,使得人们将其用于团簇组装型材料的制备过程时有了更大的自由度.
负离子光电子能谱;密度泛函;超卤素;团簇组装型材料
0 引言
在团簇的形成过程中,某些特定大小和电子组成的团簇相对于其他团簇来说,化学性质和物理性质都特别稳定,并且在与其他原子或团簇发生相互作用时能够保持结构上的稳定性与完整性,同时它们常常表现出与元素周期表中某个元素或某族元素相似的性质,这类团簇被称为超原子.团簇科学的一个重要任务便是探索和寻找高稳定性的团簇基元用来制备新型材料,而超原子正好符合这一条件.超原子的反应活性与团簇的大小和组成都有很大关系,对超原子反应活性的研究可以揭示催化反应机理,从而指导设计有特定功能的催化剂;其次,对超原子的研究为人们从原子水平设计材料奠定了理论基础,为制造和发展新的功能性纳米材料开辟了新的途径.
超原子的研究最早可以追溯到1981年Gutsev等[1]对超卤素的理论研究,他们提出了一个很简单的公式MX(n+1)/m来表示某一类超卤素.在这个公式中,n代表金属原子的最大原子价,m代表X原子的原子价,X原子的数目直接影响所组成的团簇是否可以被称为超卤素.随后,Boldyrev和他的研究小组[2-5]通过一系列的具体理论研究验证了他们所提出的这个公式的正确性.Al13是超原子的一个典型代表.1989年Castleman研究小组对铝团簇反应活性的研究[6]结果表明,Al13-,Al23-和Al37-团簇在与O2反应前后质谱峰强度没有变化,而其他的Aln-团簇在与O2反应后质谱峰强度有显著下降,这说明Al13-,Al23-和Al37-团簇的稳定性好,不与O2发生反应.这3个团簇所对应的电子数为40,70和112,刚好符合jellium模型的闭壳层结构.他们的研究结果表明,团簇的稳定性不仅可以由质谱峰的强度反映,也可以由它们的反应活性来反映.接下来对掺杂了其他元素的铝团簇的理论和实验研究[7-10]进一步为jellium模型电子结构的正确性提供了依据.从1992年开始,围绕着①有闭壳层电子结构的团簇有较强的稳定性以及②有较强稳定性的团簇应该是闭壳层的电子结构这两个问题,很多研究小组展开了一系列的实验和理论研究[11-19].在1994年Khanna等的理论研究[13]得到了Al13的电子亲和能大约为3.7 eV,与卤素中的Cl原子的电子亲和能接近,并且Al13和K原子可以像KCl一样组合形成离子键,Al13被认为是超卤素,表现出与卤素相似的性质,这是Al13第一次被正式认定是超原子,随后Castleman小组的研究为这一概念提供了更多的实验依据[20-21].在这之后,越来越多的研究小组开始关注超原子,掀起了研究超原子的热潮[22-24].另一个里程碑式的发现[25]是超原子Al7可以像原子一样与不同元素原子结合时表现出多价态的性质.
2007年Wang研究小组利用光电子能谱和理论计算相结合的方法对BO-和BO2-团簇进行了研究[26],结果表明,BO2-的电子亲和能比Cl原子的电子亲和能还要大,达到了4.46 eV,同时BO2的中性结构差1个电子达到满壳层,所以BO2具备了成为超卤素的条件.本文通过负离子光电子能谱和密度泛函的理论计算研究BO2与Cu,Na原子的相互作用,所形成团簇的电子特性以及BO2团簇的超卤素性质.
1 Cu(BO2)n-(n=1~2)系列负离子团簇
1.1 实验和理论计算方法
实验在带有激光溅射团簇源的飞行时间质谱—磁瓶式光电子能谱仪上完成[27].简单地说,532 nm的激光溅射到在平面内做平动兼转动的Cu/B合金靶(样品直径13 mm,Cu与B物质的量比为50∶1)的表面.产生的等离子体与背景压力为405.3 kPa的氦气碰撞冷却形成团簇,单脉冲激光能量大约为10 mJ.样品表面的氧化足以产生所需的Cu(BO2)n-系列负离子团簇,所以不需要再在载气管道中加入氧气.产生的负离子团簇由飞行时间质谱仪进行检测分析,然后由质量门分别选出其中的CuBO2-和Cu(BO2)2-团簇被第二束激光脱附进行光电子能谱的检测.这里使用了两种脱附激光:266 nm和193 nm,单脉冲激光能量控制在0.1~0.2 mJ.实验中采用Cu-光电子能谱进行定标,光电子能谱仪的分辨率为40 meV/eV.
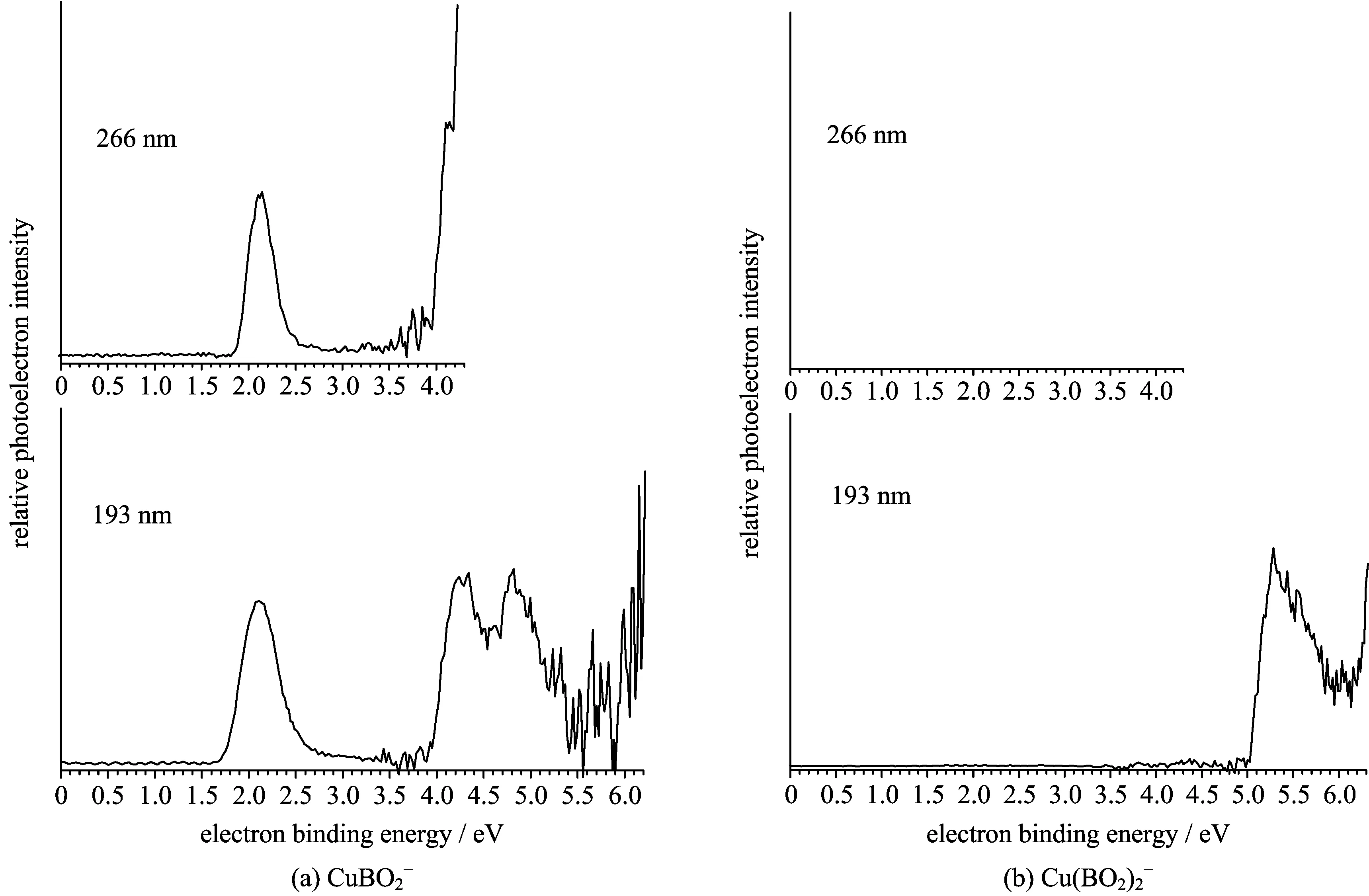
图1 在266 nm和193 nm条件下测得的CuBO2-和Cu(BO2)2-负离子团簇的光电子能谱
理论计算采用密度泛函理论中的B3LYP方法对负离子和中性团簇进行了结构优化,其中B和O原子采用了全电子基组6-311++G(3df),Cu原子采用了Lanl2dz基组,整个计算过程在Guassian09[28]软件包上完成.优化过程中,放开了对称性限制,通过频率分析确定得到的结构是否为局部稳定点.所有的能量都考虑了零点能校正.最后根据得到的每个结构的能量计算相应的垂直脱附能(vertical detachment energy, VDE)和绝热脱附能(adiabatic detachment energy, ADE).其中,垂直脱附能定义为负离子团簇和相同构型下中性团簇的能量差,而绝热脱附能定义为负离子团簇和以此负离子团簇构型作为初始构型优化得到的中性构型的能量差,一般情况下,绝热脱附能又等于中性团簇的电子亲和能(electron affinity, EA).
1.2 结果与讨论
图1给出的是在266 nm和193 nm光子能量条件下测得的CuBO2-和Cu(BO2)2-负离子团簇的光电子能谱.光电子能谱谱峰代表的是从团簇负离子基态向对应的中性团簇基态和激发态的跃迁,所以从负离子团簇的光电子能谱可以得到负离子团簇的垂直脱附能(VDE)和绝热脱附能(ADE),表1中列出了实验测量得到的VDE,ADE值以及相应的计算值.考虑到由仪器分辨引起的谱峰的展宽,我们通过以下经验方法来确定ADE的实验值:画一条与谱图中第一个峰左侧大部分重合的直线,直线与谱峰基线的交点所对应的数值再加上仪器分辨率的一半作为团簇的ADE实验值(具体操作中所加的值为0.03 eV).

表1 实验上测量得到的以及理论计算的Cu(BO2)n-团簇的ADE和VDE值
【注】 实验值的不确定度为±0.08 eV

图2 CuBO2-负离子及相应中性团簇的结构
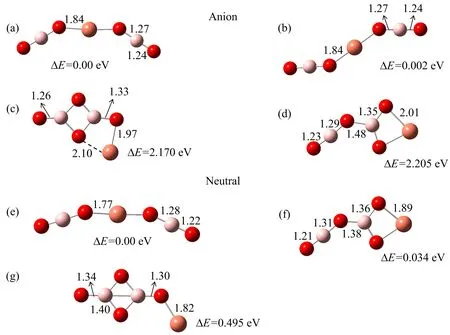
旁边的数字表示各异构体之间的相对能量差
首先我们来看CuBO2-团簇的光电子能谱.CuBO2-的193 nm的光电子能谱(图1(a),193 nm)中,第一个峰出现在2.12 eV附近,并且强度很强,随后在4.21和4.79 eV附近有两个较宽的没有完全分开的谱峰.测量得到的CuBO2-团簇的ADE和VDE实验值分别为1.90和2.12 eV.由Koopman定理可知,光电子能谱中的前两个峰分别对应中性团簇的最低非占据轨道(LUMO)和最高占据轨道(HOMO),因此中性团簇的HOMO-LUMO能量间隙也就是光电子能谱中前两个峰的能量差.所以从CuBO2-团簇的光电子能谱得到CuBO2中性团簇的HOMO-LUMO能量间隙大约为2.1 eV.HOMO-LUMO能量间隙可以反映该物种的化学惰性,能量间隙越大,该特种的化学惰性就越强.CuBO2团簇的HOMO-LUMO能量间隙为2.1 eV,这就说明CuBO2中性团簇是比较稳定的,所以它们可能是非金属性的,并且应该不具有化学反应活性.
Cu(BO2)2-团簇的绝热脱附能很大,因此在266 nm条件下无法得到有关Cu(BO2)2-团簇的光电子能谱谱峰的信息,在Cu(BO2)2-团簇的193 nm的光电子能谱中也只看到了5.28 eV附近的一个峰.根据光电子能谱,我们测得Cu(BO2)2-团簇的ADE和VDE实验值为5.07和5.28 eV.
为了更好地分析实验现象以及理解Cu(BO2)n-团簇的结构和电子特性,接下来我们结合理论计算分析Cu(BO2)n-负离子团簇以及相应中性团簇的几何结构,负离子团簇的ADE,VDE值以及中性团簇的EA值.Cu(BO2)n-(n=1~2)负离子团簇以及对应中性团簇的能量较低的几个异构体分别列于图2和图3中.
图2中的(a)和(b)给出的是CuBO2-负离子团簇的基态以及它的中性团簇的基态几何构型.从图可以看出,CuBO2-负离子团簇基态的几何构型与相应的中性团簇基态的几何构型相似,只是键长有些许不同.在这两个结构中,BO2基元仍然是直线型的结构,保持了结构上的完整性与稳定性.我们还对CuBO2-负离子团簇以及相应的中性团簇进行了NBO(natural bond orbital)电荷分析,结果表明,在CuBO2中性团簇中,Cu原子带电荷+0.87e,Cu与BO2之间有很明显的电荷转移现象发生,电荷从Cu原子流向BO2,Cu与BO2以离子键的方式结合形成CuBO2中性团簇.当增加一个电荷形成负离子团簇CuBO2-的时候,所增加的电荷的大部分(89%)都加在了Cu原子上.计算得到的CuBO2-团簇的VDE值为2.17 eV,CuBO2中性团簇的EA值为2.04 eV,与实验上观测得到的VDE(2.12 eV)和ADE(1.90 eV)值非常地接近.CuBO2-负离子团簇几何构型与相应中性团簇几何构型的相似性说明负离子团簇的ADE值也就是中性团簇的EA值.
接下来分析Cu(BO2)2团簇的负离子和中性结构.这个团簇的负离子和中性结构所对应的能量较低的几个异构体列于图3中.Cu(BO2)2-负离子的基态构型是一个顺式的C2v结构,Cu原子位于中间,与两个BO2基元以Cu—O键相连.相应的反式构型与顺式构型在能量上只相差0.002 eV,而其他的包含了B2O4基元的异构体(图3(c)和3(d))与基态的顺式结构相比能量要高很多,至少在2.0 eV以上.顺式与反式构型之间的能垒小于0.024 eV,这个值在程序计算的误差范围内.Cu(BO2)2中性团簇(4e构型)与负离子团簇的基态构型相似,而且包含了B2O4基元的异构体能量(图3(f))与基态构型能量相比,只相差了约0.03 eV.
Cu(BO2)2团簇最大的一个特点是电子亲和能EA.计算得到的Cu(BO2)2-负离子团簇的VDE和ADE值分别为5.41和5.31 eV,与实验值符合得很好,并且负离子基态构型与中性基态构型相似,所以计算得到的负离子的ADE值也就是中性团簇的EA值,也就是说,Cu(BO2)2的电子亲和能为5.31 eV,这是在我们得到的所有Cun(BO2)m(n,m=1,2)团簇中电子亲和能最大的团簇.Cu(BO2)2由中心的金属原子Cu与两个BO2超卤素基元相连,它的电子亲和能比相应的超卤素BO2的电子亲和能(4.46 eV)还要大,因此Cu(BO2)2团簇也是超卤素.Cu(BO2)2团簇较大的电子亲和能可以从NBO电荷分布来理解.在Cu(BO2)2中性团簇中,Cu原子带有+1.38e,而两个BO2基元分别带有-0.69e,Cu原子与BO2以离子键方式结合.而在形成Cu(BO2)2-负离子团簇时,NBO电荷分析显示额外的电子分布在整个Cu(BO2)2团簇上,58%位于Cu原子,剩下的42%分布在BO2上.
2 NaBO2(H2O)n-(n=0~4)系列负离子团簇
2.1 实验和理论计算方法
实验过程与前面类似,样品换成硼酸钠,背压为405.3 kPa的He气掺杂水蒸气通过脉冲阀进入团簇源,与溅射产生的等离子体碰撞,并使之冷却成簇.NaBO2-(H2O)n团簇的质谱峰可根据谱峰的同位素分布进行确认.其中n=0~4的团簇经质量门选质,然后减速,接着飞入脱附区.在脱附区,团簇与第二束激光(532 nm)相互作用,负离子吸收光子能量后脱附电子,被脱附的电子向周围空间飞出,因受到“磁瓶”约束而进入向上的螺线管飞行筒,最后由位于飞行筒末端的MCP(micro-channel plate)检测.
所有的计算都在Gaussian09程序包上完成.NaBO2-(H2O)n(n=0~4)负离子团簇以及相应的中性团簇首先用密度泛函中的B3LYP方法和6-311+G**基组进行结构优化,整个优化过程中去除对称性限制,并通过频率计算以确定得到的构型是局部最稳定结构.为了更精确地反映水分子对团簇的影响,在此基础上采用CCSD/6-311+G**方法进一步进行单点能的计算.最后的能量采用零点能校正.
2.2 结果讨论
图4给出的是在532 nm光子能量条件下测得的NaBO2-(H2O)n(n=0~4)的光电子能谱图.我们还尝试了用193 nm光子进行检测,但是没有得到其他的谱峰信息,因此这里只给出了532 nm条件下的谱图.实验中得到的各负离子团簇的绝热脱附能ADE和垂直脱附能VDE值列于表2中.
首先来看NaBO2-团簇的光电子能谱图(图4),NaBO2-谱图中有一个强峰,中心位置在1.00 eV,这个峰对应的是从NaBO2-负离子基态向NaBO2中性基态的跃迁.实验测得的NaBO2团簇的ADE和VDE值分别为0.93和1.00 eV.前面已经说过,在193 nm的光子能量(6.424 eV)下测得的NaBO2-光电子能谱中没有看到第二个峰,这说明NaBO2的HOMO-LUMO能量间隙至少为5.4 eV.NaBO2-(H2O)和NaBO2-(H2O)2的光电子能谱与纯的NaBO2-团簇的光电子能谱非常相似,只不过是随着水分子数目的增加相应团簇的谱峰逐渐向高电子束缚能方向有所移动,NaBO2-(H2O)谱峰位置相对移动了0.23 eV,NaBO2-(H2O)2谱峰位置相对移动了0.38 eV.这个现象是NaBO2-溶剂化效应的一种体现,说明NaBO2-正在与水分子发生相互作用.

X峰来源于Na-BO2-(H2O)n构型的异构体,X′峰来源于Na(H2O)n…BO2-构型的异构体
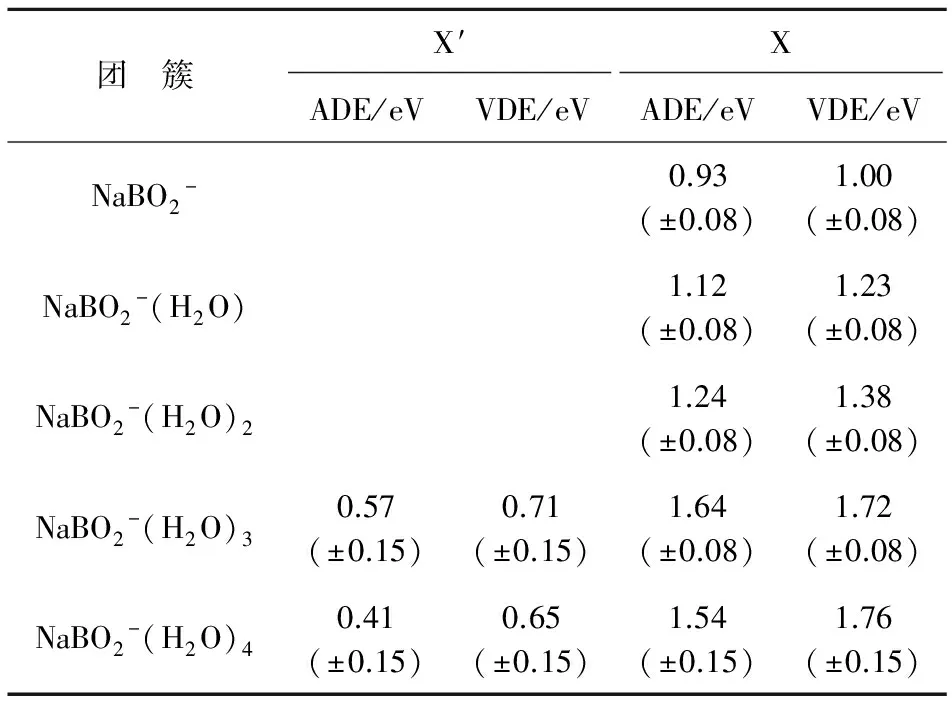
表2 实验上得到的NaBO2-(H2O)n(n=0~4)团簇的VDE和ADE值
当NaBO2-结合的水分子的数目继续增加以后,光电子能谱有了很明显的改变.在NaBO2-(H2O)3的光电子能谱中,在0.71 eV附近有一个很宽的峰(X′),随后有一个强峰,中心位置位于1.72 eV(X).同样的,在NaBO2-(H2O)4的光电子能谱中也有相似的两个峰出现,分别位于0.65 eV(X′)和1.76 eV(X)附近.标注为X的峰的峰型与纯的NaBO2-的谱峰很相似,并且也是随着水分子数目的增加,谱中心位置相应地往高电子束缚能方向移动.而NaBO2-(H2O)3和NaBO2-(H2O)4的光电子能谱中的X′峰的出现,可能是由于NaBO2-受水分子作用的影响而导致电子结构改变所产生的,这在后面将有所讨论.
理论计算得到的NaBO2-(H2O)n(n=0~4)团簇以及相应中性团簇的能量较低的异构体分别列于图5和6中,其中最左边对应的是能量最低的结构.每个结构所对应的ADE和VDE值列于表3中,为了方便比较,表3中还给出了ADE和VDE的实验测量值.
NaBO2-的最稳定结构0A是一个线型结构,Na原子与O=B=O的末端的氧原子相连.计算得到的0A结构的ADE和VDE值也与实验测量值一致.其他的异构体0B和0C在能量上比0A结构高至少4 eV,并且计算得到的ADE和VDE值也与实验值不符合,因此我们排除了0B和0C结构在实验中出现的可能.

图5 NaBO2-(H2O)n(n=0~4)团簇的能量较低的异构体
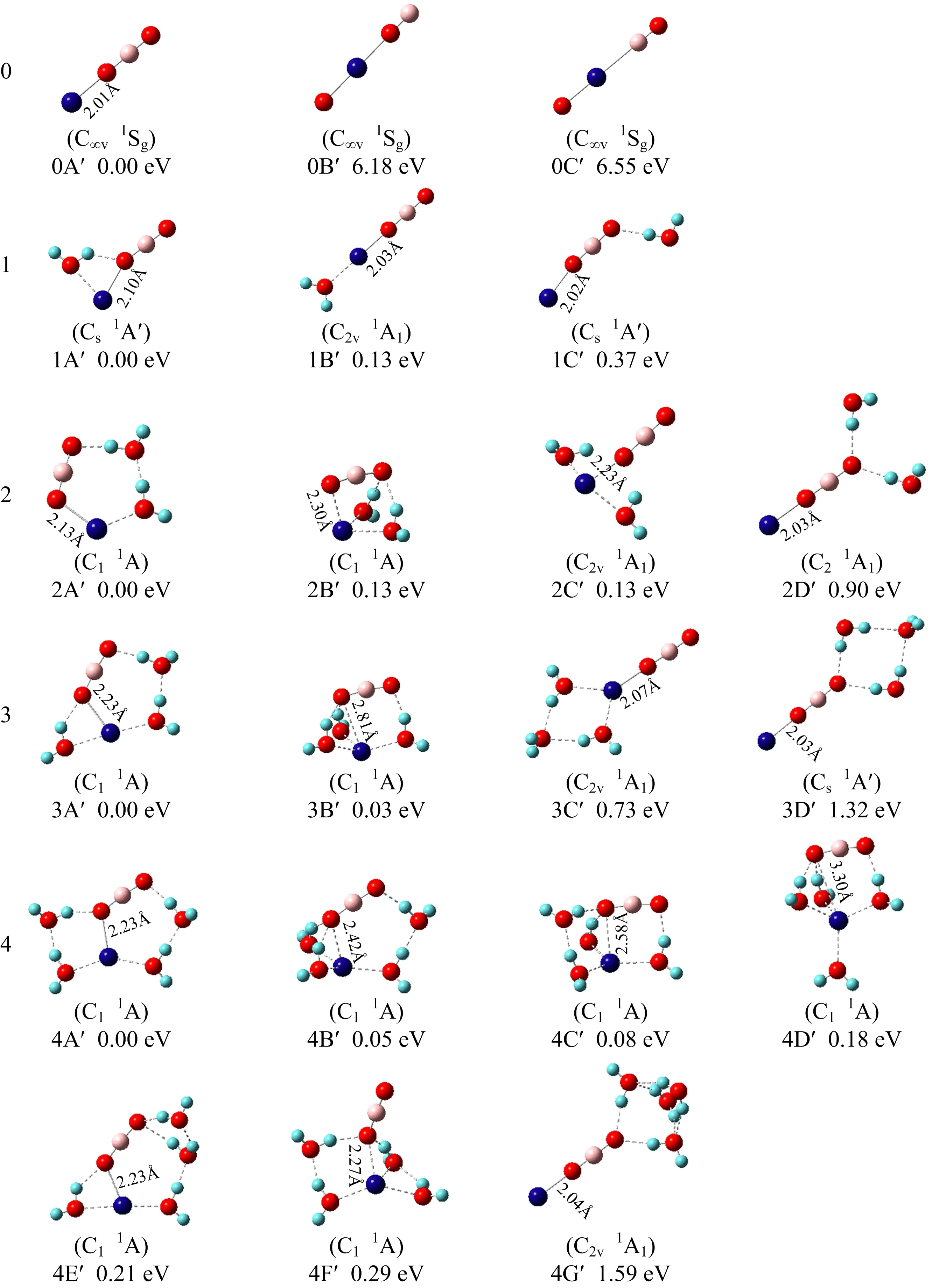
图6 NaBO2(H2O)n(n=0~4)团簇的能量较低的异构体

表3 NaBO2-(H2O)n(n=0~4)团簇的异构体间的相对能量以及计算得到的各结构的ADE和VDE值
NaBO2的中性团簇也是一个线型结构,但是Na—O键相对于负离子团簇来说稍微短一些.这与以前的计算结果是一致的[29].中性团簇的密立根电荷分布显示,正电荷集中在Na原子上,负电荷集中在BO2上,具体分布为(Na)0.946+(OBO)0.946-,在得到电子形成负离子团簇的时候,所加的电荷主要分布在Na原子上,这也与碱金属盐的电荷分布规律一致.另外,NaBO2中性团簇的电偶极矩也很大,达到了13.1 D,这个数值与典型的离子化合物的电偶极矩相当,比如NaCl(9.0 D), KCl(10.3 D), NaBr(9.1 D), 以及KBr(10.6 D).而且测量得到的NaBO2团簇的电子亲和能约为0.93 eV,与碱金属盐的电子亲和能也相似,如NaF (0.520 eV), NaCl (0.727 eV), NaBr(0.788 eV),以及NaI (0.865 eV)[30].因此,NaBO2可以被认为是超卤素盐,表现出与卤盐相似的性质.
用同样的方法我们还找到了与实验对应的含水团簇的结构(表3中的黑体标注).NaBO2-(H2O)n系列团簇可以分为Na-BO2-(H2O)n和Na(H2O)n…BO2-两种类型的结构,NaBO2-(H2O)和NaBO2-(H2O)2的最稳定结构是Na-BO2-(H2O)n类型,在这个类型中,水分子易于与NaBO2末端的O原子形成氢键.而对于NaBO2-(H2O)3和NaBO2-(H2O)4团簇,两种类型的结构同时存在,并且各结构在能量上非常接近.其中能量最低的结构对应的是Na(H2O)n…BO2-类型,这个类型中水分子易于和Na原子相连.NaBO2-(H2O)n(n=1~4)团簇的光电子能谱中高束缚能处的峰对应的是Na-BO2-(H2O)n类型的结构,比如1A,2B,3D,4E结构.Na-BO2-(H2O)n类型的结构中,Na-(OBO)键受水分子的影响很小,NaBO2-基元也没有明显的变化,因此1A,2B,3D,4E结构的光电子能谱与纯的NaBO2-光电子能谱非常相似,只不过随着水分子数目的增加,峰的位置逐渐往高电子束缚能方向移动.NaBO2-(H2O)3和NaBO2-(H2O)4光电子能谱中低束缚能处的峰对应的是Na(H2O)n…BO2-类型的结构,这个类型结构中,水分子与Na原子和BO2-基元都有很强的作用.Na-(OBO)键明显拉长,整个NaBO2-也不再保持直线型结构.
对于NaBO2(H2O)n的中性团簇,水分子易于与Na+相结合,这可能是由于水分子中的O原子有孤对电子,而Na原子刚好带正电荷的原因,这样一来,水分子可以通过与Na+分享孤对电子来使整个团簇达到稳定结构.而那些与NaBO2末端O原子相连的结构比如说1C′, 2D′, 3D′和4G′则相对不稳定,这与以前报道的NaCl(H2O)n体系[11]中Na+-(H2O)相互作用强于Cl--(H2O)相互作用是一致的.此外,在NaBO2-(H2O)n负离子团簇中,随着水分子数目的增加,水分子与Na原子之间的相互作用会有所加强.因此,水分子数目增加以后,NaBO2-(H2O)n负离子团簇的最稳定结构与相应的中性团簇的最稳定结构相似.
4 结论
我们利用光电子能谱和密度泛函理论研究了Cu(BO2)n-(n=1~2)和NaBO2-(H2O)n(n=0~4)系列团簇的构型、电子结构、垂直脱附能、绝热脱附能和相应中性团簇的电子亲和能.结果表明,BO2在与Na,Cu原子相互作用形成团簇的过程中表现出与卤素相似的特性,同时NaBO2-团簇的水溶性也与卤盐相类似,随着水分子数目的增加,会相继出现紧密离子对(CIP)和溶剂隔离的离子对结构(SSIP)类型的结构.这些都证实了BO2团簇的超卤素性质.另外,在实验中发现Cu(BO2)2-团簇的电子亲和能比相应的卤素和超卤素的电子亲和能还要大,为了区别,我们定义其为“长链二级超卤素”.长链二级超卤素由3种元素原子组成,在一定程度上提高了材料制备过程中的自由度.
References)
[1] Gutsev G L, Boldyrev A I. DVM-Xα calculations on the ionization potentials of MXk+1-complex anions and the electron affinities of MXk+1“superhalogens” [J]. Chem Phys, 1981, 56(3): 277-283.
[2] Boldyrev A I, von Niessen W. The first ionization potentials of some MHk+1-and M2H2k+1-anions calculated by a Green’s function method [J]. Chem Phys, 1991, 155(1): 71-78.
[3] Gutsev G L, Boldyrev A I. The way to systems with the highest possible electron affinity [J]. Chem Phys Lett, 1984, 108(3): 250-254.
[4] Gutsev G L, Boldyrev A I. Theoretical estimation of the maximal value of the first, second and higher electron affinity of chemical compounds [J]. J Phys Chem, 1990, 94(6): 2 256-2 259.
[5] Boldyrev A I, Simons J. Is TeF82-the MXn2-dianion with the largest electron detachment energy (5 eV) [J]. J Chem Phys, 1992, 97(4): 2 826-2 827.
[6] Leuchtner R E, Harms A C, Castleman A W, Jr. Thermal metal cluster anion reactions: Behavior of aluminum clusters with oxygen [J]. J Chem Phys, 1989, 91(4): 2 753-2 754.
[7] Leuchtner R E, Harms A C, Castleman A W, Jr. Aluminum cluster reactions [J]. J Chem Phys, 1991, 94(2): 1 093-1 101.
[8] Leskiw B D, Castleman A W, Jr. The interplay between the electronic structure and reactivity of aluminum clusters: Model systems as building blocks for cluster assembled materials [J]. Chem Phys Lett, 2000, 316(1/2): 31-36.
[9] Wagner R L, Vann W D, Castleman A W, Jr. A technique for efficiently generating bimetallic clusters [J]. Rev Sci Instrum, 1997, 68(8): 3 010-3 013.
[10] Harms A C, Leuchtner R E, Sigsworth S W, et al. Gas-phase reactivity of metal alloy clusters [J]. J Am Chem Soc, 1990, 112(14): 5 673-5 674.
[11] Khanna S N, Jena P. Assembling crystals from clusters [J]. Phys Rev Lett, 1992, 69(11): 1664.
[12] Khanna S N, Jena P. Atomic clusters: Building-blocks for a class of solids [J]. Phys Rev B, 1995, 51(19): 13 705-13 716.
[13] Khanna S N, Jena P. Designing ionic solids from metallic clusters [J]. Chem Phys Lett, 1994, 219(5/6): 479-483.
[14] Liu F, Mostoller M, Kaplan T, et al. Evidence for a new class of solids. First-principles study of K(Al13) [J]. Chem Phys Lett, 1996, 248(3/4): 213-217.
[15] Ashman C, Khanna S N, Liu F, et al. (BAl12)Cs:mA cluster-assembled solid [J]. Phys Rev B, 1997, 55(23): 15 868.
[16] Ashman C, Khanna S N, Pederson M R. Reactivity of AlnC clusters with oxygen: Search for new magic clusters [J]. Chem Phys Lett, 2000, 324: 137-142.
[17] Rao B K, Khanna S N, Jena P. Designing new materials using atomic clusters [J]. J Cluster Sci, 1999, 10(4): 477-491.
[18] Ashman C, Khanna S N, Pederson M R, et al. Al7CX (X=Li-Cs) clusters: Stability and the prospect for cluster materials [J]. Phys Rev B, 2000, 62(24): 16 956-16 961.
[19] Ashman C, Khanna S N, Pederson M R. Electron attachment and dynamics of alkali atoms in Al13X (X=Li-Cs) clusters [J]. Phys Rev B, 2002, 66(19): 193 408.
[20] Bergeron D E, Castleman A W, Morisato T, et al. Formation of Al13I-: Evidence for the superhalogen character of Al13[J]. Science, 2004, 304(5667): 84-87.
[21] Bergeron D E, Roach P J, Castleman A W, et al. Al cluster superatoms as halogens in polyhalides and as alkaline earths in iodide salts [J]. Science, 2005, 307(5707): 231-235.
[22] Kumar V, Kawazoe Y. Hund’s rule in metal clusters: Prediction of high magnetic moment state of Al12Cu from first-principles calculations [J]. Phys Rev B, 2001, 64(11): 115-405.
[23] Kumar V, Kawazoe Y. Metal-encapsulated icosahedral superatoms of germanium and tin with large gaps: Zn@Ge12and Cd@Sn12[J]. Appl Phys Lett, 2002, 80(5): 859-861.
[24] Zheng W J, Thomas O C, Lippa T P, et al. The ionic KAl13molecule: A stepping stone to cluster-assembled materials [J]. J Chem Phys, 2006, 124(14): 144 304.
[25] Reveles J U, Khanna S N, Roach P J, et al. Multiple valence superatoms [J]. Proc Natl Acad Sci USA, 2006, 103: 18 405-18 410.
[26] Zhai H J, Wang L M, Li S D, et al. Vibrationally resolved photoelectron spectroscopy of BO-and BO2-: A joint experimental and theoretical study [J]. J Phys Chem A, 2007, 111(6): 1 030-1 035.
[27] Xu H G, Zhang Z G, Feng Y, et al. Vanadium-doped small silicon clusters: Photoelectron spectroscopy and density-functional calculations [J]. Chem Phys Lett, 2010, 487: 204-208.
[28] Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian 09[CP].Wallingford CT: Gaussian Inc, 2009.
[29] Ramondo F, Bencivenni L, Sadun C. Ab initio SCF study of the BO2ion and the NaBO2and HBO2molecules [J]. J Mol Struct (Theochem), 1990, 209(1/2): 101-109.
[30] Miller T M, Leopold D G, Murray K K, et al. Electron affinities of the alkali halides and the structure of their negative ions [J]. J Chem Phys, 1986, 85(5): 2 368-2 375.
Experimental and theoretical investigation of the superhalogen character of BO2cluster
FENG Yuan1,2, XU Hongguang2, HOU Gaolei2, KONG Xiangyu2
(1.School of Electronics and Information Engineering, Hefei Normal University, Hefei 230601, China;2.State Key Laboratory of Molecular Reaction Dynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China)
Superhalogens exhibit special stable characters and electronic properties, and are suitable to be used as building blocks in designing cluster assembling materials. BO2is only one electron short to electronic shell closing and has a high electron affinity, 4.46 eV, larger than that of halogen atoms, so BO2can be considered as a superhalogen. In order to confirm this, the interactions between BO2and Cu, Na atoms as well as microscopic salvation of NaBO2cluster were intensively studied through anion photoelectron spectroscopy (PES) and density functional theory (DFT) calculation. The studies show that BO2moiety still retains its linear structure as the bare BO2cluster and behaves as a superhalogen. On the other hand, the microscopic solvation of NaBO2in water is similar to that of halogen salts. NaBO2appears as contact ion pair (CIP) structure at first, and then there is a significant change in the photoelectron spectra of NaBO2-(H2O)nclusters starting fromn=3, corresponding to the transition from contact ion pair (CIP) structure to solvent-separated ion pair (SSIP) structure. Besides, with an electron affinity of 5.07 eV, which is larger than that of its BO2superhalogen building-block, Cu(BO2)2can be classified as a hyperhalogen. Since the hyperhalogen contains three different elements, it has much more freedom in designing materials.
anion photoelectron spectroscopy; density functional theory; superhalogens; cluster-assemble materials
0253-2778(2013)05-0369-10
O643.1
A
10.3969/j.issn.0253-2778.2013.05.004
Feng Yuan, Xu Hongguang, Hou Gaolei, et al. Experimental and theoretical investigation of the superhalogen character of BO2cluster[J]. Journal of University of Science and Technology of China, 2013,43(5):369-378.
冯源,许洪光,候高垒,等. BO2团簇超卤素特性的实验和理论研究[J]. 中国科学技术大学学报,2013,43(5):369-378.
2012-11-29;
2013-03-20
国家自然科学基金青年科学基金(21201052),安徽高校自然科学研究重点项目(KJ2013A224),合肥师范学院人才科研启动基金(2012rcjj01)资助.
冯源(通讯作者),女,1985年生,博士/讲师. 研究方向:团簇物理. E-mail: yfeng@iccas.ac.cn
猜你喜欢
现代农村科技(2023年3期)2023-04-14
材料研究与应用(2022年5期)2022-11-07
浙江林业科技(2022年1期)2022-02-20
科学与生活(2021年3期)2021-11-10
山西大同大学学报(自然科学版)(2021年2期)2021-05-04
中学化学(2017年1期)2017-03-17
中学生理科应试(2016年12期)2017-01-07
重庆工商大学学报(自然科学版)(2015年10期)2015-12-28
原子与分子物理学报(2015年3期)2015-11-24
液晶与显示(2014年2期)2014-02-28
