
Al-Mg-Si合金Pre-β″与β″析出相的界面原子键络与性能
2013-12-14 07:44高英俊黄礼琳黄创高
中国有色金属学报 2013年4期
高英俊,韦 娜,黄礼琳,黄创高
(广西大学 物理科学与工程技术学院,南宁 530004)
Al-Mg-Si 合金由于具有低密度、较高强度和优良力学性能,已被广泛应用于车辆和飞机结构件等领域[1-2]。最新研究发现[3-5],在有过剩 Si的Al-Mg-Si合金,时效温度为200~300 ℃范围时,合金微结构的析出相变顺序按以下方式进行[5]:

该合金在时效过程中产生的析出相对合金的性能有着不同的影响[5-8]。目前,对 pre-β″、β″、β′和U1、U2相开展的研究,主要集中在实验上对该相的晶体结构的测定和析出相稳定性的研究。在理论上,尽管有人用第一原理计算其价电子结构[9-10],但计算其内部原子成键,目前还很少有相关报道。因此,研究和掌握这些析出相的内部原子键特征,对优化合金材料的性能有非常重要的意义。
基于价键理论[11]和能带理论建立的固体与分子经验电子理论(EET)[12],以及改进的界面TFD理论[13],在处理复杂体系合金的电子结构方面提供了一个简捷实用的经验方法,使得对该合金宏观性能的研究可以追溯到原子成键的电子结构层次。高英俊研究小组[14-21]之前已对Al-Mg-Si合金的GPZ、U1、U2和β相的电子结构进行了系统地计算和研究,本文作者则是将EET与改进的TFD方法结合,对该合金的pre-β″和β″析出相内部原子间的价电子成键及其与基体界面间形成的界面键络特征进行研究,从原子成键角度分析该析出相的原子键络特点及其对合金性能的影响。
1 相空间结构模型
pre-β′相是β′相的析出前相,也是属于 GP 区的一个后续演化相,研究该相的价电子结构可以更好地认识合金从GP区结构转化为β′相的机理。文献[9, 22]给出pre-β′相晶胞结构为单斜晶胞(C2/m),其晶格常数为a=1.460 nm,b=0.405 nm,c=0.640 nm,β=105°,与基体共格,取向关系为:(001)Al//(010)β″,[10]Al//[001]β″,[230]Al//[100]β″。根据上述给出的晶格常数和空间对称性,用专门的晶体结构绘图软件可以画出该相的晶胞结构如图1(a)所示。
该结构单元中,拥有Mg1、Mg2、Mg33类Mg原子和Si1、Si2、Si33类Si原子,它们在晶胞中具有各自的对称位置。例如,除了Mg1原子外,其它Mg原子都是分布在XZ平面以及与该平面平行的面上,并且与XZ平行的面上Mg原子分布也与XZ面上的原子分布相同。之所以把Mg原子分为3类,是因为Mg1、Mg2、Mg3原子的周围环境不相同,类似地Si1、Si2、Si3情况也一样。
β″相是 Al-Mg-Si合金的峰值时效的最重要析出相,该相的晶体结构[5-7]也是一个单斜晶胞,分子式近似为Mg5Si6,与基体共格[6]。晶格常数为a=1.516 nm,b=0.405 0 nm,c=0.674 nm,β=105.3°,其晶胞结构如图1(b)所示。β″相沿a、c轴方向,与基体的取向关系为[22](001)Al//(010)β″, [310]Al//[001]β″,[230]Al//[100]β″。
从β″相的晶体结构看与pre-β″相的结构相似,其原子的空间分布也具有相似的对称性,并且都只是在XZ面和与其平行的面上分布,但其原子位置还是有明显差别。从图1(b)可以看出,Mg1原子的位置就与pre-β″中的不同,pre-β″中的Mg1原子分布在底面的棱上,而β″中的Mg1占据的是顶角的位置,

图1 pre-β″相和-β″相的晶胞结构Fig.1 Cell structures of pre-β″(a) and β″(b) phases
其他原子的具体坐标位置也有差别。详细的原子空间位置坐标见文献[18]。因此,可知这两个相虽有一定联系,但也有差别,正是这些结构差别使得这两个相对合金的影响也有所不同。
2 计算方法
2.1 EET方法简介
固体中原子的价电子结构在EET理论中是指该固体中原子所处的状态以及原子形成共价键的键络分布。按照EET[12-13]理论,原子的共价电子是分布在连接最近邻、次近邻, 以及s近邻原子的键上。u和v原子的各键上共价电子对数(即键级nα)由下列原子键距公式表示:
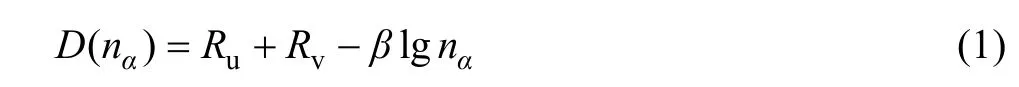
式中:D(nα) 是共价键距,Ru、Rv分别是u和v原子的单键半距,β为参量,计算中参数β的数值按文献[12-13]中的公式确定。晶胞内的共价电子数满足下述方程关系:

式中:k1、k2分别为晶胞中u、v原子的个数,nuc、nvc分别为u、v原子的共价电子数。Ⅰαs为nα键级的等同键数,各等同键数的选取可依照文献[12]给出的方法来确定。由于各晶胞的结构已确定,实验晶格常数文献[4-5, 22]已给出,因此,运用键距差(BLD)方法[12]建立最强键nA方程,并参见文献[16, 23-24]的求解步骤,联立(1)、(2)等方程组,逐个计算各晶胞中原子成键的价电子结构, 并依据BLD判据确定原子的杂阶状态ε。
2.2 改进的TFD理论简介
文献[13]定义了异相界面电子结构,指出异相界面电子结构除包括相界面两侧平面上的键络电子分布外,还包括相界面两侧平面上的平均共价电子密度ρ(hkl)、ρ(uvw) 和面电子密度的相对差值分数Δρ,以及使界面电子密度在一级近似下保持连续的原子状态组数σ。相界面电子结构的计算是在空间电子结构计算的基础上进行的。
对于(hkl)α//(uvw)β异相界面电子结构的计算,首先要求出在α和β相空间中,符合键距差判别条件ΔDnα≤0.05 nm的原子状态,在此基础上,应用“界面上的电子密度连续”的边界条件(在一级近似下,以Δρ<10%来判断界面电子密度的连续性.当Δρ<10%时,把电子密度定义为连续或连续性较好;当Δρ>10%时,界面电子密度认为是偏离连续或连续性较差),得到异相界面的原子键络的强度分布。然后计算该异相界面两侧平面(hkl)和(uvw)上的平均共价电子密度ρ(hkl)、ρ(uvw),界面处电子密度的相对差值百分数Δρ,其具体计算表达式由改进的TFD理论[11]给出如下:

改进的 TFD理论给出的异相界面电子结构的物理意义:异相界面处的电子密度ρ愈高,其原子键络密度就越大,界面结合得就越牢固;而异相界面处的相对电子密度差Δρ愈小,界面上的电子密度连续性就愈好,界面原子键络匹配得就越好,界面畸变能就越低,界面畸变应力也愈小。连续性状态组数σ值越大,晶界抵抗外力作用的能力越强,破坏该界面就越难,就越不容易断裂;反之,界面畸变应力就愈大,界面畸变能就越高,界面就越不稳定.当畸变应力大到超过临界值时,则界面电子密度的连续性遭到破坏,这时将伴随在界面处析出新相或在宏观上出现裂纹或断裂。界面电子密度的连续性的好坏,实质上是由于点阵原子键络畸变和缺陷而导致的结果,直接影响到材料的性能好坏。
2.3 结合能计算
2.3.1 结合能理论计算公式
文献[12]给出了金属化合物u和v原子结合的结合能计算公式和详细说明:
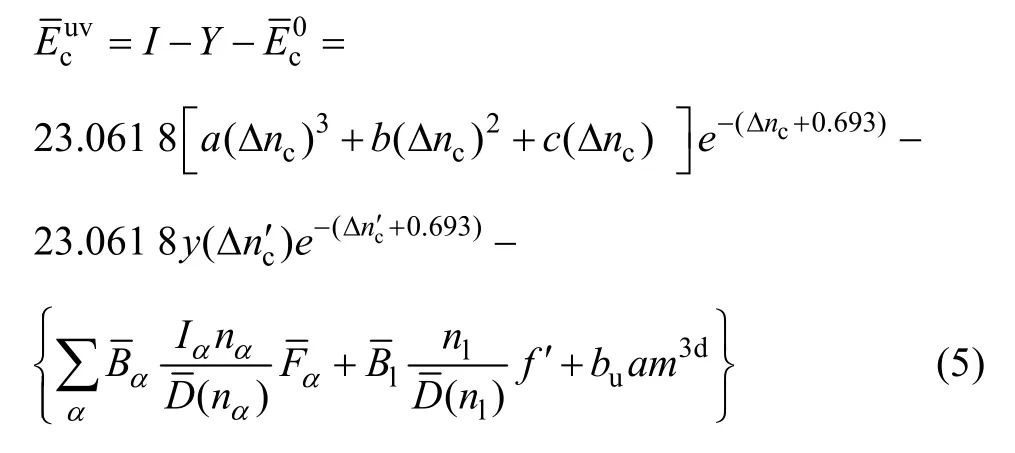
式中:Ⅰ为晶体中不同种类的原子间相互成键时多提供电子的原子的类离化能,Δnc为多提供电子的原子“输出”的电子数,23.061 8是把eV转换为4.184 0 kJ/(g·mol)时的变换当量;e-(Δnc+0.693)表示衰减因子;a、b、c是参数,由电离能实验数据确定;Y为当不同种类原子间相互成键时,少提供共价电子的原子类亲合能;y则是原子的亲合能; Δnc’为“输入”电子数(即晶体中偏移的电子数);为共价电子的屏蔽系数;u、v为形成α键的原子,自由电子的屏蔽系数m、n为分子式中包含的u和v的原子数,bu、bv为 u、v元素晶体结合能的屏蔽系数;杂化键结合因子具体表达式为式(5)中其余符号可参阅文献[12]。
2.3.2 结合能实验计算公式
文献[12]给出了结合能实验计算公式为

式中:是结合能的实验计算值;H0为实验测量f的晶体umvnwt的形成热(以吸热为正);EcE、EcE、EcEuvw分别为u、v 和w原子的结合能;式中元素晶体结合能取绝对值;m、n和l分别为u、v和w的原子数;公式中所有单位都取4.184 0 kJ/(g·mol)。详细说明见文献[12]。

表1 结合能的理论计算公式中的相关参数值[12]Table1 Parameters of cohesive energy in theoretical calculation[12]
2.3.3 结合能计算公式的相关参数
pre-β″和β″相的结合能计算公式(5)和(6)中的相关参数的取值,分别列在表1和表2。表1中m3d为磁性电子参数,nT为原子总价电子数,nl为晶格电子数,fα为原子成键能力。由于Al-Mg-Si合金是非磁性金属,故在本研究中,不需考虑m3d的作用。Hf为实验测得的晶体结构形成的生成热焓。
通常直接从实验测量出晶体的结合能是很困难的。为了与实验值比较,通常是通过实验测量该相的生成焓,然后用式(6)计算该相的实验值。若实验计算值与理论计算值差别不大,说明结合能的计算结果是合理的。

表2 结合能实验计算公式中的相关参数值[12, 25]Table2 Parameters of cohesive energy in experimental calculation formula[12, 25]
2.4 界面能计算方法
根据推广的Becker模型[26],把形成异相界面的两个异相分别记为α相和β相,计算时把基体相记为α相,β″和Per-β″相记为β相,把 Al、Mg、Si原子分别记为A、B、C原子,则α//β界面属于α(A-B)//β(B-C)类型的界面,这时,异相界面能的计算公式[27]可表示为


式中:Zint为界面上的平均键密度,等于界面上的原子密度NS与面配位数ZR的乘积,可用穿过界面的键数与界面面积的比值来计算;cαB、cCβ分别为α相中B原子和β相中C原子的浓度;Δc为界面两侧溶质的浓度差;εα和εβ分别为α、β相中AB原子组成ABAB的键的键能;εα、εα、εβ、εβ、εβ分别为α、AABBAABBCCβ相中AA、BB、CC原子组成的键的键能;εα、εβABAC和εβ为α和β相中AB、AC和BC原子组成的键的BC键能。
3 结果与分析
3.1 原子键络结果
根据EET方法,按照上节2.1所介绍的计算步骤,计算出 pre-β′和β′原子键络强度结果如表3 和表4 所列。由表3 和表4 可见,pre-β′和β′相的原子键络最强键都是 Si—Si键,但β′相的 Si—Si最强键比 pre-β′′相的Si—Si最强键要强,nα达到0.784 21。由键能强度Eb也可看出β′相的 Si—Si键络骨架要比 pre-β′相的Si—Si键络骨架要强。因此,β′相对合金的强化作用要比pre-β′相要显著。之所以形成强的Si—Si键络骨架,是因为Si原子有较强的相互作用。在作者研究小组之前,蒙特卡洛方法研究[17,21]Al-Mg-Si合金的微结构形成机理时,发现在早期新相形成过程中,容易形成 Si团簇,成为形成 GPZ 和pre-β′和β′相的前期晶胚。从表3和表4还可以看出,Si—Si键要比Mg—Si键要强得多。在pre-β′相中,Si原子都处于5杂阶,而β′相中,部分Si原子处于更高的杂阶,即第6阶,全为共价电子,数目为nc=4。因此,β′相的共价键原子更多,形成的共价键更强,所以,β′相对合金的强化作用更显著。这与文献[2]的实验结果吻合。关于基体α(Al)的原子键络,主要键强为 Al—Al键,基体中含有少量的Mg和Si原子,这里的Al—Mg和Al—Si键是较弱的,这些键的强度在作者研究小组 已发表的论文有介绍[18-20]。例如,Al—Al键强为 0.208 57,Al—Mg 键强为 0.205 37 和Al—Si键强为 0.154 38,相比本文得到的 pre-β′和β′相的 Si—Si键强,就弱很多。因此,pre-β′和β′相的析出是能够起强化作用的。
3.2 界面电子结构
基体Al的界面电子结构已在前期工作中计算出,详见文献[14-16, 18-21, 23-24]。pre-β″和β″相与α(Al)基体界面电子结构,按照EET和改进的TFD理论方法,用计算机扫描满足基体α(Al)、pre-β″和β″相空间价电子结构中键距差ΔDnα<0.005 nm 条件的界面电子密度保持连续的解,即键络强度、Δρ<10%的原子状态组数σ,最小电子密度差Δρ的结果列在表5和表6。
从表5的计算结果可见,pre-β″相的界面电子结构中,其平均界面电子密度为9.913 2 nm-2,界面电子密度差为9.363%,状态组数为σ=17 200,这些信息表明了Al-Mg-Si合金的亚稳相pre-β″与基体的界面电子密度差在一级近似下保持好的连续性,异相界面结合得较好。参看空间电子结构与界面电子结构的原子杂阶变化,可见Si1、Si2、Si3原子状态由杂阶ε=5都跃迁到了ε=6杂阶,但是由于Si原子的尺寸在不同的杂阶没有变化,半径都是0.119 nm,所以对pre-β″相结构的作用保持不变,而Mg2原子的杂阶状态由ε=4降至ε=2杂阶,原子半径由0.125 21 nm增加到0.125 73 nm,原子尺寸稍变大。由于主键由 Si原子键组成,因此Mg2的变化对晶格畸变的影响并不明显。这些都说明此界面连续性还是比较好的。
从表6中可看出,α//β″面上的平均电子密度差较小,Δρmin只有0.110 2%,说明它与基体形成的界面结合匹配较好,畸变应力较小,界面较稳定,有利于合金界面的强化;符合Δρ<10%的原子状态组数σ较多,达到σ=11 194,说明该相界面电子密度的连续性好,相界面抵抗外力作用的能力就强,也就是说该相界面在外力作用下界面不易被破坏,能较好地避免出现裂纹或断裂。参看空间电子结构与界面电子结构原子的杂阶,可见Si1、Si3原子杂阶都有了变化,Si1原子杂阶从第6阶降到了第5阶,Si3原子从第3阶跃迁到了第5阶,说明界面的形成使得这三类原子杂阶趋于一致,界面结构更匹配,也使得该界面更稳定。但是由于 Si原子的尺寸在不同杂阶都保持相同半径,都为0.117 nm,所以影响不大,而Mg原子的杂阶都有了变化,原子尺寸稍变大。但由于主键由Si原子键组成,因此Mg的变化对晶格畸变的影响并不是很大。这些都说明此界面连续性还是比较好的,对合金的强化是有利的。

表3 pre-β′的原子键络强度Table3 Atomic bonding of pre-β′ phase

表4 β′原子键络强度Table4 Atomic bonding of β′ phase

表5 pre-β′(010)//Al(001)界面原子成键Table5 Interface atomic bonding of pre-β′(010)//Al(001)
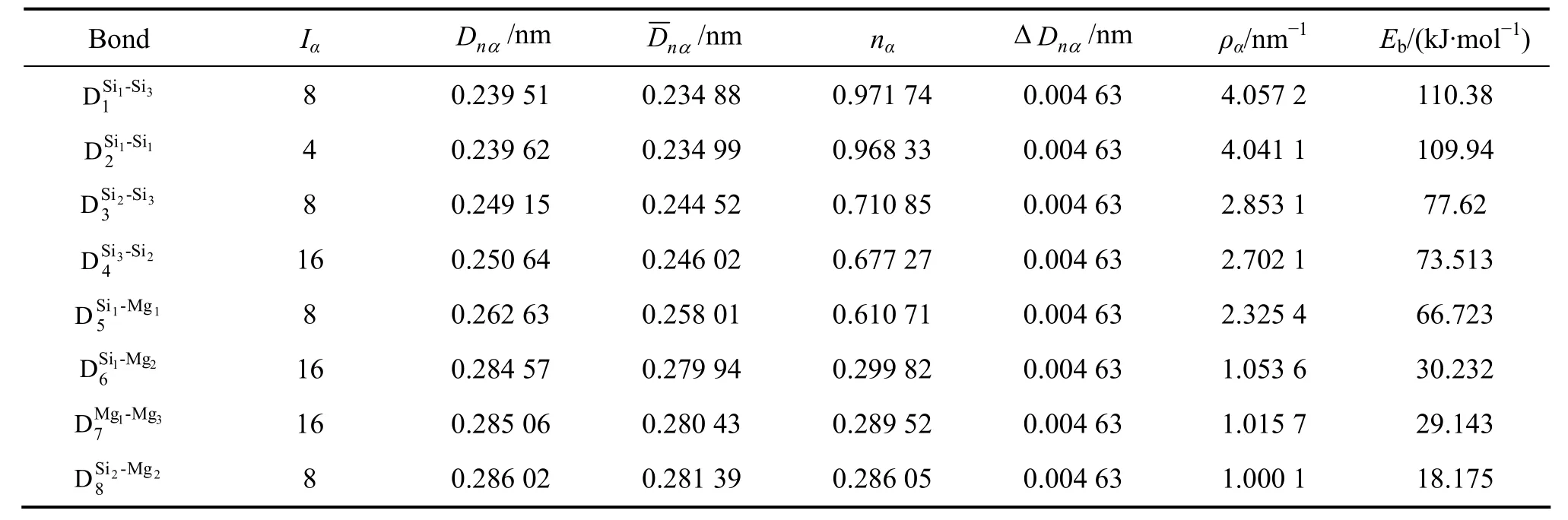
表6 β′(010)//Al(001)界面原子成键Table6 Interface atomic bonding of β′(010)//Al(001)
3.3 结合能和界面能
利用式(5)和(6)计算得到的pre-β″和β″相的理论结合能ECth和实验结合能ECexp的结果,以及利用公式(7)计算得到的界面能结果列在表7。

表7 界面能和结合能结果Table7 Results of interface and cohensive energy
由表7的结果可知,pre-β′相的结合能计算值与实验值相差小于10%,因此可以认为计算的结合能是合理的,而文献用第一原理计算得到的结果与实验计算值相差 20%,偏离实验值较大。由计算得到的β′′相结合能实验值与理论值进行对比,可见计算值与实验值相差 10.5%,由此可以认为该相计算结果是合理的。β′结合能为-3.757 kJ/mol,比其前析出相 pre-β′′的结合能-3.325 kJ/mol大,表明β′相更稳定。由于β′′相的键络要比 pre-β′相的键络要强,因此其熔点和硬度都应高于 pre-β′相。表7给出了计算的 Al(001)//pre-β′相的界面能为 65.45 mJ/m2,Al(001)//β′相的共格界面能为127.63 mJ/m2。可见,pre-β′相的界面能小,需要的形核功小,该相形核阻力较小。按界面能大小对合金析出顺序的影响,可知该相较易析出。相对而言,β′相的界面能较高,界面能大,表明界面面积不易增加,该相形核长大较难。因此,该相析出长大较晚,符合析出相序列的实验结果。
4 结论
1)在Al-Mg-Si 系合金时效过程中,析出相pre-β″、β″ 的最强共价键nA为 Si—Si键,强度分别为0.636 47和0.784 21,都比基体的共价键nA=0.208 57大2倍,最强键键能值分别为69.298和83.521 kJ/mol,两相的结合能值分别为-3.325和-3.757 kJ/mol,这些因素对合金的影响在宏观上表现为对基体的强度增大,提高基体的硬度。
2) pre-β″、β″这两个相与基体间界面的电子密度差Δρ都较小,其值分别 9.363%、0.11025%,说明这两个相与基体形成的界面都保持较好的连续性,这种连续性有利于该相晶粒的晶界迁移和长大。这在两个析出相与基体的界面上的平均电子密度ρ分别为9.913 2和10.386 nm-2,这些结果说明界面结合较好,有利于合金的界面强化。由此可以推断,这两个析出相能够有效地提高合金的强度,与实验所报道的情况相符合。
3) 由于pre-β′相的界面能较小,需要的形核功小,因此,界面结构更稳定。界面能小的相易于析出,按界面能大小对合金析出顺序的影响,可知该相较易析出。相对而言,β′相的界面能较高,界面能大,说明界面面积不易增加,该相形核长大较难。因此,该相析出长大较晚,符合析出相序列的实验结果。
[1]HIROSAWA S, SATO T.Nano-Scale clusters formed in the early state of phase decomposition[J].Mater Sci Forum, 2005,475/479: 357-360.
[2]杨文超, 汪明朴, 盛晓菲, 张 茜.轨道交通车辆6005A合金板材料时效析出及硬化行为研究[J].金属学报, 2010, 46(12):1481-1487.YANG Wen-chao, WANG Ming-pu, SHENG Xiao-fei, ZHANG Qian.Study of the ageing precipitation and hardening behavior of 6005A alloy[J].Acta Met Sin, 2010, 46(12): 1481-1487.
[3]VISSERS R, van HUIS M A, JANSEN J, ZANDBERGEN H W,MARIOARA C D, ANDERSEN S J.The crystal structure of theβ′ phase in Al-Mg-Si alloys[J].Acta Materialia, 2007, 55:3815-3823.
[4]FROSETH A G, HOIER R.Bonding in MgSi and Al-Mg-Si compounds relevant to Al-Mg-Si alloys[J].Physical Review,2003, B67: 224106.
[5]ANDERSEN S J, MARIOARA C D, FRØSETH A, VISSERS R,ZANDBERGEN H W.Crystal structure of the orthorhombic U2-Al4Mg4Si4precipitate in the Al-Mg-Si alloy system and its relation to theβ′ andβ" phases[J].Materials Science and Engineering A, 2005, 390: 127-138.
[6]TSAO C S, CHEN C Y.Precipitation kinetics and transformation of metastable phases in Al-Mg-Si alloys[J].Acta Materialia,2006, 54: 4621-4631.
[7]ANDERSEN S J, MARIOARA C D, VISSERS R, FROSETH A,ZANDBERGEN H W.The structural relation between precipitates in Al-Mg-Si alloys[J].Materials Science and Engineering A, 2007, 444: 157-169.
[8]MATSUDA K, SAKAGUCHI Y, MIYATA Y.Precipitation sequence of various kinds of metastable phases in Al-Mg-Si alloy[J].J Mater Sci, 2000, 35: 179-189.
[9]RAVI C, WOLVERTON C.First-principles study of crystal structure and stability of Al-Mg-Si-(Cu) precipitates[J].Acta Materialia, 2004, 52: 4213-4227.
[10]VAN M A, CHEN J H.Phase stability and structural relations of nanometer-sized, matrix-embedded precipitate phases in Al-Mg-Si alloys in the late stages of evolution[J].Acta Materialia, 2006, 54: 2945-2955.
[11]鲍林 L.化学键本质[M].上海: 上海科学技术出版社, 1996:393.PAULING L.The nature of chemical bonds[M].Shanghai:Shanghai Science and Technology Press, 1996: 393.
[12]张瑞林.固体与分子经验电子理论[M].长春: 吉林科学技术出版社, 1993: 1-250 ZHANG Rui-lin.Empirical electron theory in solids and molecules[M].Changchun: Jilin Science and Technology Press,1993: 1-250.
[13]刘志林, 李志林, 刘伟东.界面电子结构与界面性能[M].北京: 科学出版社, 2002: 23.LIU Zhi-lin, LI Zhi-lin, LIU Wei-dong.Electron structure and properties of interface[M].Beijing: Science Press, 2002: 23.
[14]高英俊, 李云雯, 王态成, 黄创高.Al-Mg-Si合金强化作用的键分析[J].轻金属, 2005, 316: 55-58.GAO Ying-jun, LI Yun-wen, WANG Tai-cheng, HUANG Chuang-gao.Analysis of bond in Al-Mg-Si alloy for strengthening[J].Light Metals, 2005, 315: 55-58.
[15]高英俊, 王庆松, 王 娜.Al-Mg-Si合金GPZ的原子键络与强化作用[J].矿冶工程, 2006, 26(5): 89-91.GAO Ying-jun, WANG Qin-song, WANG Na.Atomic bonding and strengthening effect of GP zones in Al-Mg-Si alloy[J].Mining and Metallurgical Engineering, 2006, 26(5): 89-91.
[16]高英俊, 陈华宁, 韦 娜, 文春丽, 黄创高.Al-Mg-Si合金的U1和U2相的原子成键与性能[J].中国有色金属学报, 2010,20(7): 1267-1272.GAO Ying-jun, CHEN Hua-ning, WEI Na, WEN Chun-li,HUANG Chuang-gao.Atomic bonding ofU1 andU2 phase and properties of Al-Mg-Si alloy[J].The Chinese Journal of Nonferrous Metals, 2010, 20(7): 1267-1271.
[17]张丽娜, 高英俊, 易 杰, 李建勋, 苏义勇.Si和Sc对Al-Cu-Mg合金时效初期微结构演化作用[J].中国稀土学报,2008, 26(5): 661-665.ZHANG Li-na, GAO Ying-jun, YI Jie, LI Jian-xun, SU Yi-yong,The role of Sc, Si on microstructural evolution of Al-Mg-Cu alloy[J].J Rare Earth Society, 2008, 26(5): 661-665.
[18]韦 娜.Al- Mg-Si合金析出相的价电子结构与力学性能[D].南宁: 广西大学, 2011.WEI Na, Electronic structure and mechanical properties of precipitation in Al-Mg-Si alloy[D].Nanning: Guangxi University,2011.
[19]陈华宁.Al-Mg-Si合金亚稳相的价电子结构与合金性能[D].南宁: 广西大学, 2009.CHEN Hua-ning, Atomic bonding and properties of precipitation in Al-Mg-Si alloy[D].Nanning: Guangxi University, 2009.
[20]王庆松.Al-Mg-Si合金强化机理的电子理论研究[D].南宁:广西大学, 2006.WANG Qin-song, The strengthening for Al-Mg-Si alloy studied by electron theory[D].Nanning: Guangxi University, 2006.
[21]易 杰.Al- Mg-Si-Zn合金微结构演化的计算机模拟研究[D].南宁: 广西大学, 2008.YI Jie.Computation simulation of microstructure evolution of Al-Mg-Si-Zn alloy[D].Nanning: Guangxi University, 2008.
[22]MARIOARA C D, ANDERSEN S J, JANSEN J.Atomic model for GPZ in Al-Mg-Si alloy[J].Acta Materialia, 2001, 49:321-328.
[23]高英俊, 黄创高, 莫其逢.Al-Li合金时效初期的价键分析[J].中国有色金属学报, 2005, 15(7): 1069-1074.GAO Ying-jun, HUANG Chuang-gao, MO Qi-feng.Electronic structure of Al-Li alloy under earlier ageing condition[J].The Chinese Journal of Nonferrous Metals, 2005, 15(7): 1069-1074.
[24]高英俊, 文春丽.Al-Li-Zr合金相界面的原子成键与力学性能[J].中国有色金属学报, 2011, 21(9): 2202-2208 GAO Ying-jun, WEN Chun-li.Interface atomic bonding and mechanical properties of Al-Li-Zr alloy[J].The Chinese Journal of Nonferrous Metals, 2011, 21(9): 2202-2208.
[25]饭田修一.物理学常用数表[M].张质贤,译.北京: 科学出版社, 1979: 85.IIDA S.Mathematical tables of physics[M].ZHANG Zhi-xian,transl.Beijing: Science Press, 1979: 85.
[26]BORCHERS C, BORMANN R.Determination of lowtemperature inter facial energies from a pair interaction model[J].Acta Materialia, 2005, 53: 3695-3701.
[27]文春丽.Al-Li-Cu-Mg合金相界面的价电子结构与力学性能[D].南宁: 广西大学, 2010: 10-30.WEN Chun-li.Electronic structure of interface boundary and mechanical properties of Al-Li-Cu-Mg alloy[D].Nanning:Guangxi University, 2010: 10-30.
猜你喜欢
广州化工(2022年20期)2022-12-01
大学物理(2022年9期)2022-09-28
石材(2022年3期)2022-06-01
石材(2022年3期)2022-06-01
物理通报(2020年7期)2020-07-01
测绘通报(2019年11期)2019-12-03
赤峰学院学报·自然科学版(2019年5期)2019-09-10
中国材料进展(2019年5期)2019-07-20
科教导刊·电子版(2018年13期)2018-07-31
科技创新导报(2017年19期)2017-09-13
