
WmBn(m+n≤7)团簇电子结构与光谱性质的计算研究
2013-11-19 09:27张秀荣
江苏科技大学学报(自然科学版) 2013年4期
张秀荣, 尹 琳, 陈 晨
(1.江苏科技大学 数理学院,江苏 镇江 212003)(2.江苏科技大学 材料科学与工程学院,江苏 镇江 212003)
过渡金属混合团簇具有特殊的电子结构和物理化学性质,为此成为目前团簇科学的热门课题[1-8].W合金材料,因其有很高的硬度、较好的耐磨、耐蚀性和延展性、抗高温氧化、易脱模、不粘着等特殊性质,引起了科技工作者的广泛关注[9-15].文献[16]利用电子散射衍射和映像分析技术研究了在烧结过程中WC颗粒的生长行为;文献[17]利用电沉积方法获得的Ni-W合金具有很强的延展性和抗拉强度,可以沿一个角度旋转180°而不使材料断裂.文献[18]对BmN (m=2~9)团簇的几何构型、电子结构、振动频率、自然键轨道(natural bond orbital,NBO)等性质进行了理论研究,得到了BmN (m=2~9)团簇结构的稳定性信息.文献[19]对AlBn+(n=2~10)团簇几何结构、稳定性、电子结构和成键特性进行了系统理论的研究,得到了AlBn+(n=2~10)团簇的最稳定结构,并且通过对其红外振动光谱的研究得出了硼原子间更容易成键的结论.文献[20]计算得出FeBn(n≤15)团簇基态结构中Fe的d轨道和B原子的P轨道存在着明显的杂化现象,研究表明FeB3、FeB5、FeB12和FeB15团簇较相邻团簇稳定.文献[21]对FeBn(n≤6)团簇的磁性做了系统研究,发现除了FeB5团簇外,FeBn(n≤6)团簇的总磁矩和Fe原子磁矩随团簇尺寸的增大而减小.文献[22]计算出了W6Sin0,±(n=1,2)团簇基态及亚稳态结构,发现Si-Si之间不成键,并且分析了其芳香性及磁性.文中将对WmBn团簇的电子结构和光谱性质进行研究.
1 计算方法
文中采用量子化学程序Gaussian03,在B3LYP/LANL2DZ水平上对WmBn混合团簇基态结构的电子结构和光谱性质进行了计算研究.为了寻找WmBn的基态结构,设计了WmBn(m+n≤7)团簇多种可能的几何结构,进行几何参数优化,把优化之后无虚频的结构定为稳定结构,把能量最低且没有虚频的结构定为基态结构.然后对基态结构的电子结构和光谱性质进行了计算研究.
2 结果与讨论
2.1 基态结构
图1给出了WmBn(m+n≤7)团簇的基态构型.图中颜色较深的大球为W原子,较浅的小球为B原子.通过图1可知,当只有一个W原子时,团簇的构型是平面结构,当m≥2,且m+n≥4时,除W3B团簇外,其余团簇的基态结构均为立体结构.对称性最高的是W2B3团簇,其对称性为D3h.

图1 WmBn(m+n≤7)团簇的基态结构Fig.1 Ground state structures of WmBn(m+n≤7) clusters
2.2 自然键轨道分析
文中采用自然键轨道(NBO)方法分析了WmBn(m+n≤7)团簇的自然电荷布局以及成键性质.处于稳定状态的原子,核外电子将尽可能地按能量最低原理排布,另外它们还要遵守泡利不相容原理和洪特规则.处于基态的原子中电子以1s,2s,2p,3s,3p,4s,3d,4p,5s,4d 次序排布,从而得出电子组态.自由W原子最外电子层排布为5s25p65d46s2.B原子最外电子层排布为1s22s22p1.表1列出了WmBn(m+n≤7)团簇基态结构的各轨道上的NBO电荷分布.
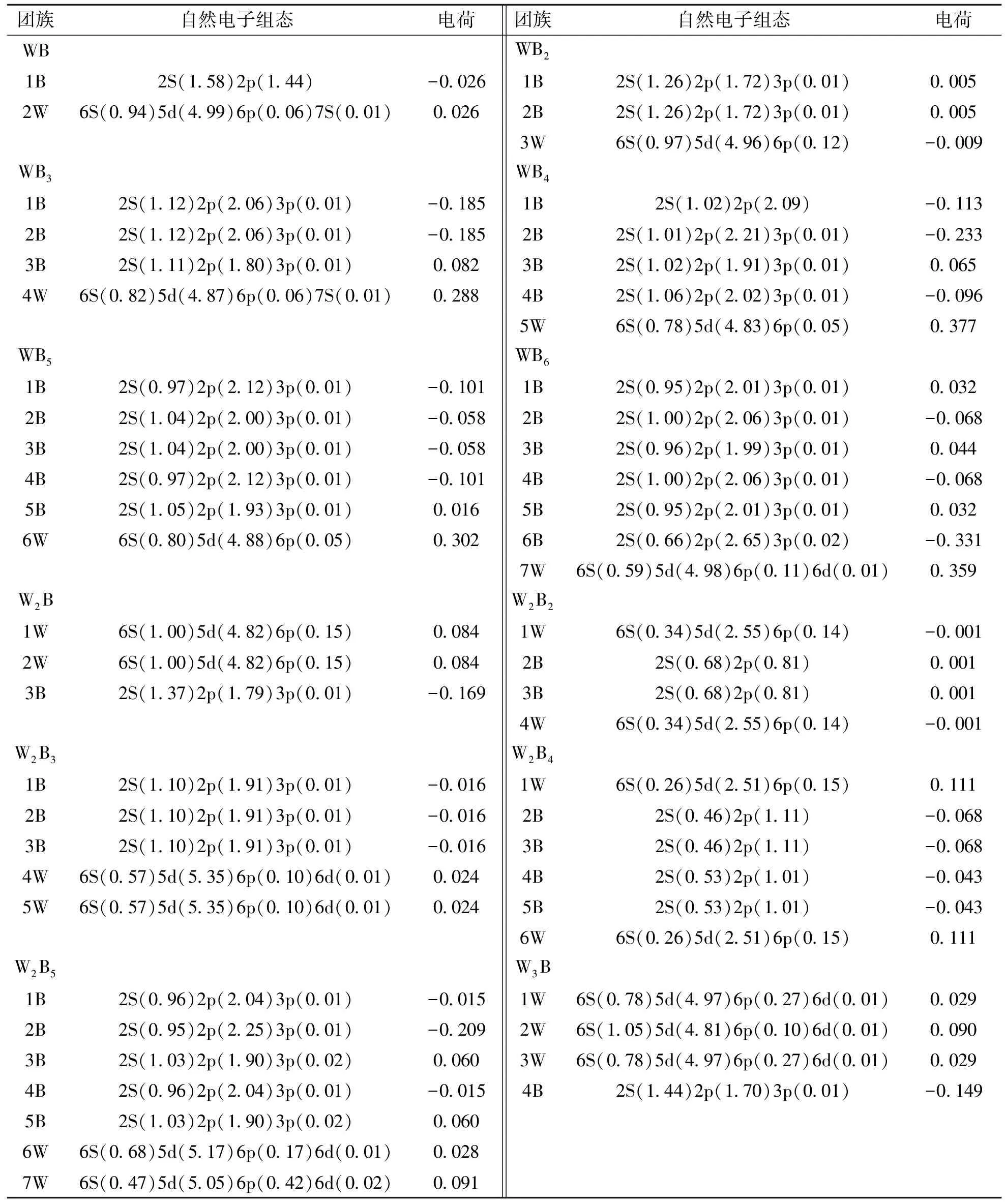
表1 WmBn (m+n≤7)团簇基态结构的自然电子组态和电荷Table 1 Natural electron configuration and atomic charge of the ground state structures of WmBn(m+n≤7) clusters
因为每个原子处在不等价的空间位置,所受到的势场不同,当一部分原子失去电荷,另一部分得到电荷就出现了电荷转移现象.从表1发现,钨原子的6s轨道上的NBO电荷分布在0.22至1.05,钨原子NBO电荷主要集中在5d轨道,分布在2.42至5.35,同时6p轨道上的NBO电荷在0.05至0.52,分布相对较少.当m<3时,部分钨原子含有6d轨道,当m≥3时,钨原子都含有6d轨道,但是电荷分布均极少,在0.01至0.02范围内.只有WB和WB3团簇中的钨原子含有7s轨道,电荷分布也较少.由上述分析以及表1看出,除W2B4、W3B2、W3B4和W4B2团簇外,钨原子的5d轨道和6p轨道得到电子,6s轨道失去电子,说明钨原子内部出现了轨道杂化现象,结论与WnNim(n+m=8)团簇[23]自然轨道电荷转移现象相吻合.硼原子中2s轨道上的NBO电荷分布在0.45至1.82,硼原子NBO电荷主要集中在2p轨道(除了WB团簇),此时NBO电荷分布在0.81至2.65,大部分硼原子含有3s轨道,但是电荷分布极少.据此以及表1可知,除W2B2,W3B4和W4B2团簇外,硼原子的2p轨道得到电子,2s轨道失去电子,硼原子内部也发生了轨道杂化,结论与WnNim(n+m=8)团簇[23]自然轨道电荷转移现象也相吻合.
深入探讨可以发现,WmBn(m+n≤7)团簇中,除了WB2和W2B2团簇外,与钨原子相邻的硼原子上的2p轨道得到电荷数目大于2s轨道失去电荷数目,而钨原子6s轨道失去电子数目大于5d,6d轨道得到的电荷数目,说明钨原子中的部分电荷转移到了硼原子,所以在两个轨道电子的相互作用的过程中,电荷从钨原子转移到硼原子,形成了W-B键.而对于WB2和W2B2团簇,W原子得到了电荷,B原子得到了电荷,即B原子2p轨道上的部分电荷转移到了W原子上,W原子变成了电荷的受体,使得W原子的6s轨道和B原子的2p轨道形成了复杂的化学键.综上所述,在W原子与B原子内部轨道之间发生了电荷转移现象,即轨道杂化现象,这些杂化轨道在原子之间相互作用形成化学键,决定了团簇的稳定性和特殊的物理化学性质.结合表1和基态结构图发现WmBn(m+n≤7)团簇中NBO电荷分布状况与团簇的对称性相关,在对称性较高的团簇中位置相同的原子,其NBO电荷分布状况也相同.如在W2B3团簇(对称性为D3h)中,1B,2B和3B的电荷分布相同均为2S(1.10)2p(1.91)3p(0.01),4W和5W原子上的NBO电荷分布均为6S(0.57)5d(5.35)6p(0.10)6d(0.01);W4B3团簇(对称性为C3V)中,对称位置上的2W和3W的NBO电荷分布状况相同,4W和5W,6B和7B的NBO电荷分布状况也分别相同.
表1同时也列出了WmBn(m+n≤7)团簇基态结构中各个原子上的净电荷分布情况.在WmBn(m+n≤7)团簇中,W原子的净电荷分布在-0.065e至0.377e,B原子的净电荷分布在-0.331e至0.083e,W原子的净电荷分布比B原子的净电荷分布范围较大一些,说明W原子比B原子对电荷调节能力要强,易与其他原子形成化学键.在W原子和B原子相互作用形的过程中,原子间发生了电荷转移,大部分W原子呈正电性,大部分B原子呈负电性,B原子从W原子得到电子,这部分W原子提供电荷,在形成团簇的过程中起主要作用.
2.3 光谱分析
一个多原子的化合物分子可能存在很多振动方式,但并不是所有的分子振动都能吸收红外光.当分子的振动不致改变分子的偶极矩时,它就不能吸收红外辐射,不具有红外活性,即红外光谱的吸收强度由振动中的偶极距变化大小决定的.分子振动(和点阵振动)与转动引起分子极化率发生变化,则产生拉曼光谱.
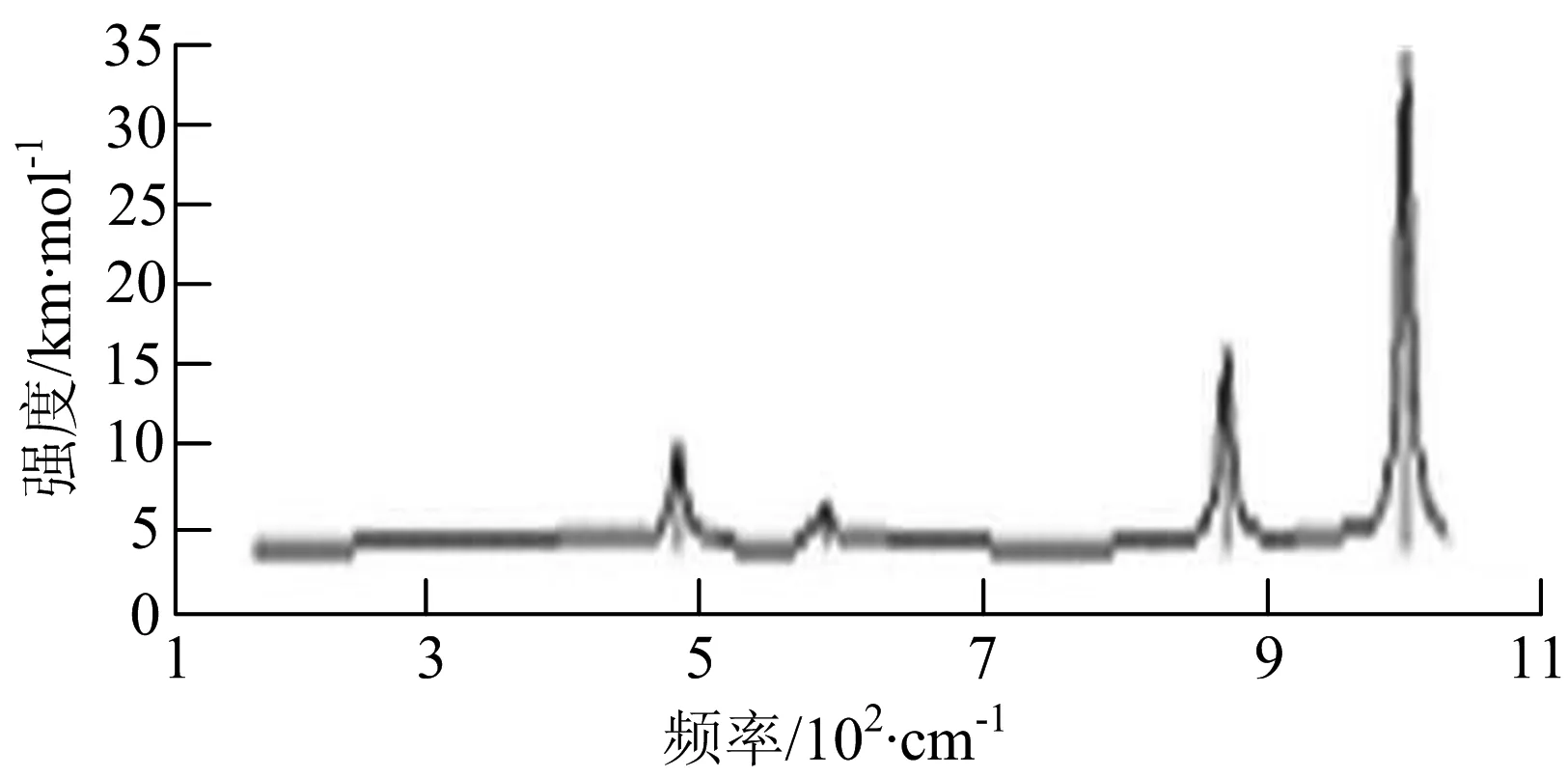
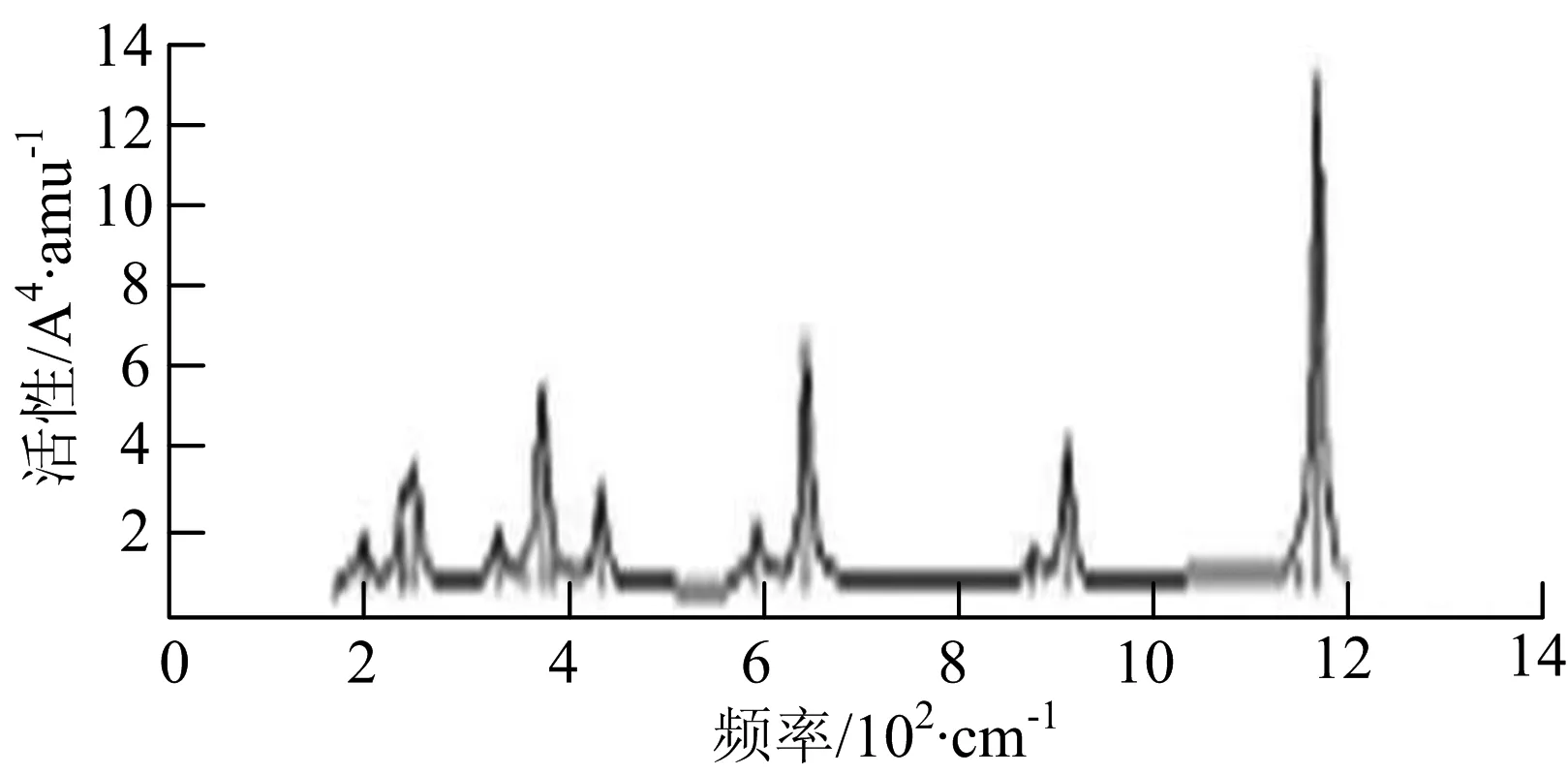
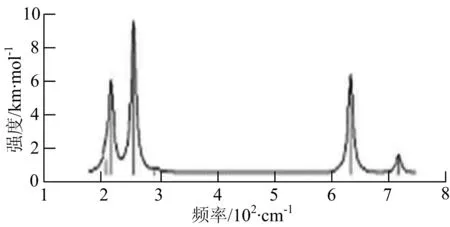
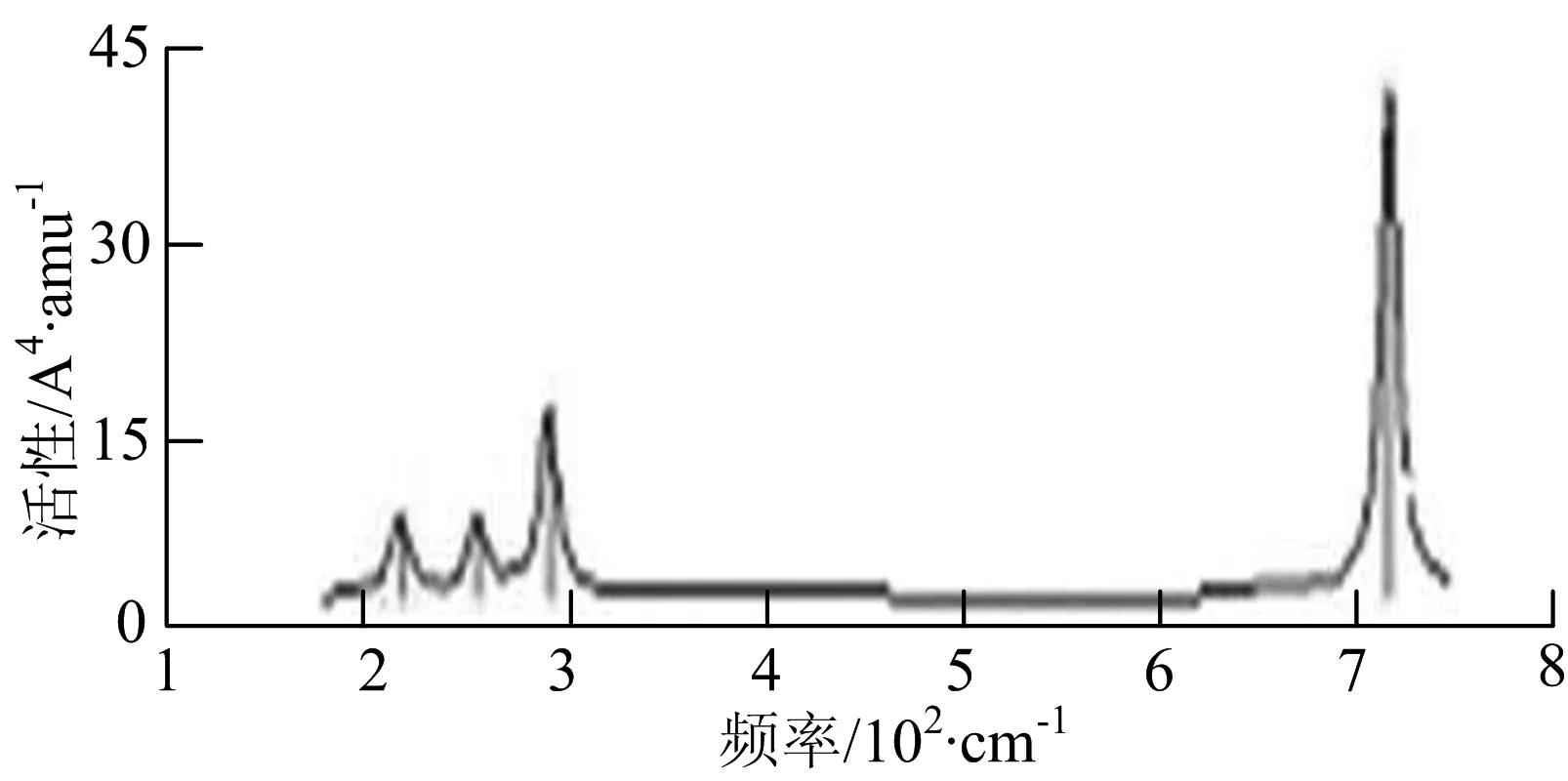
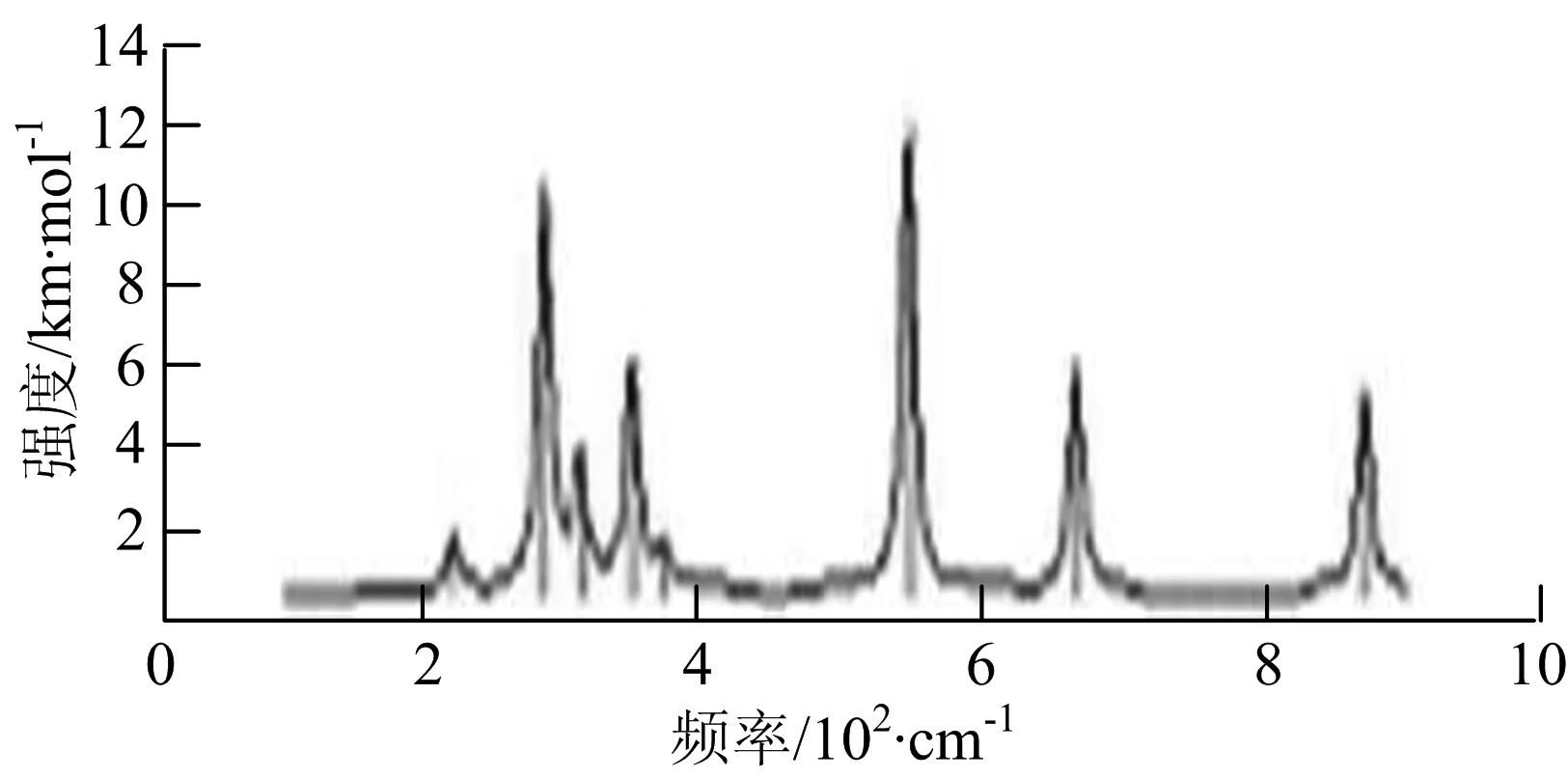
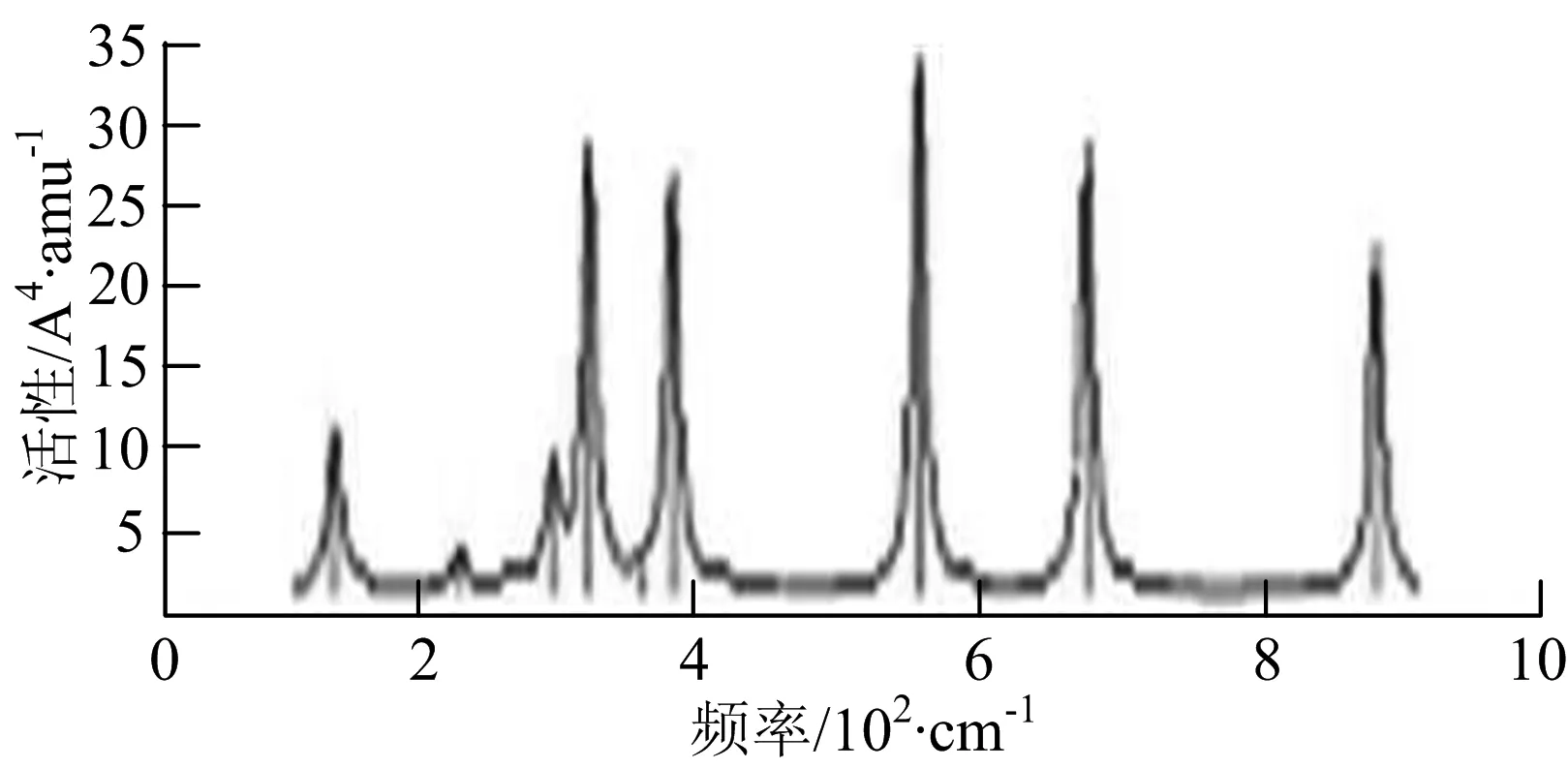
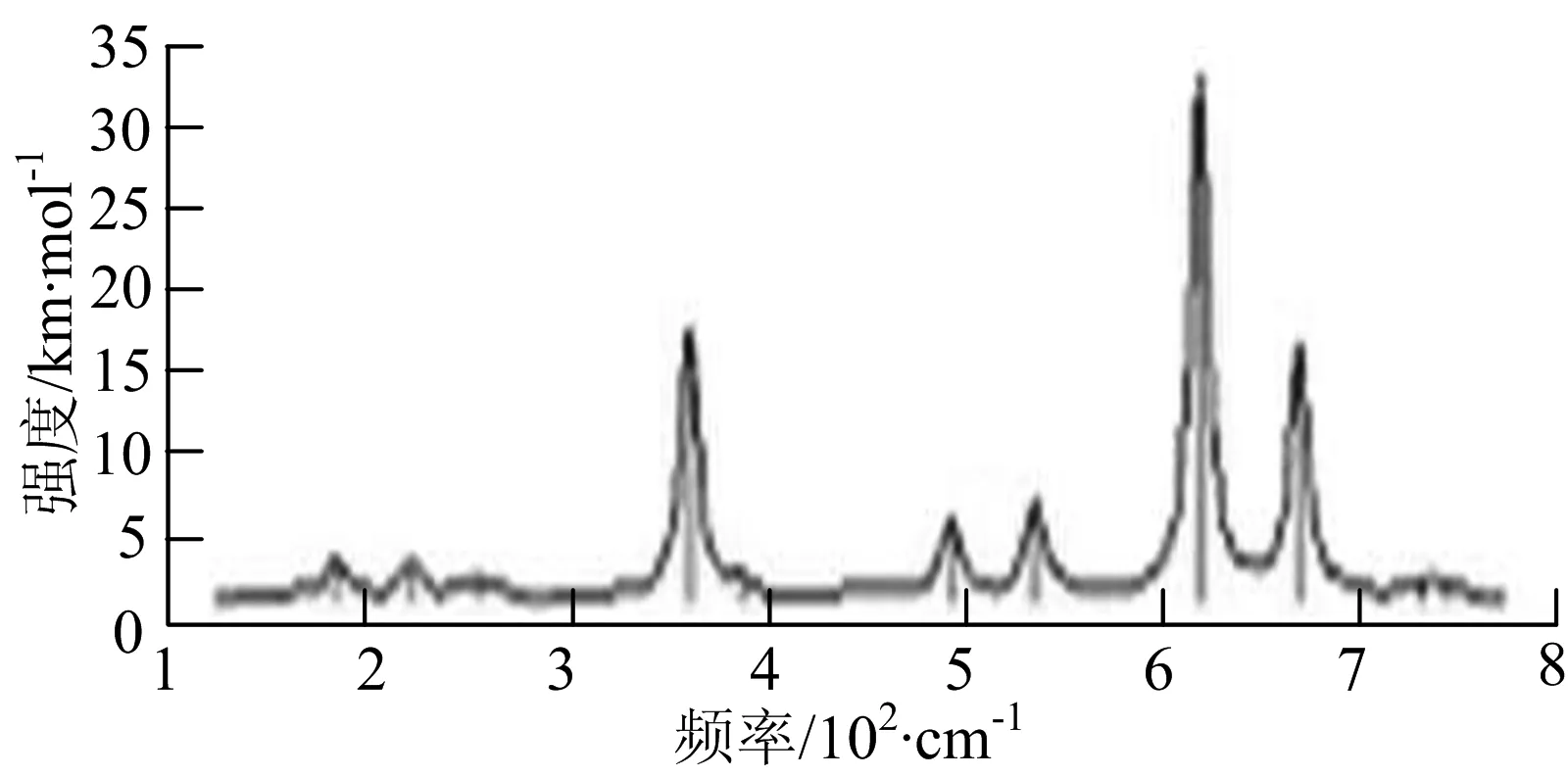
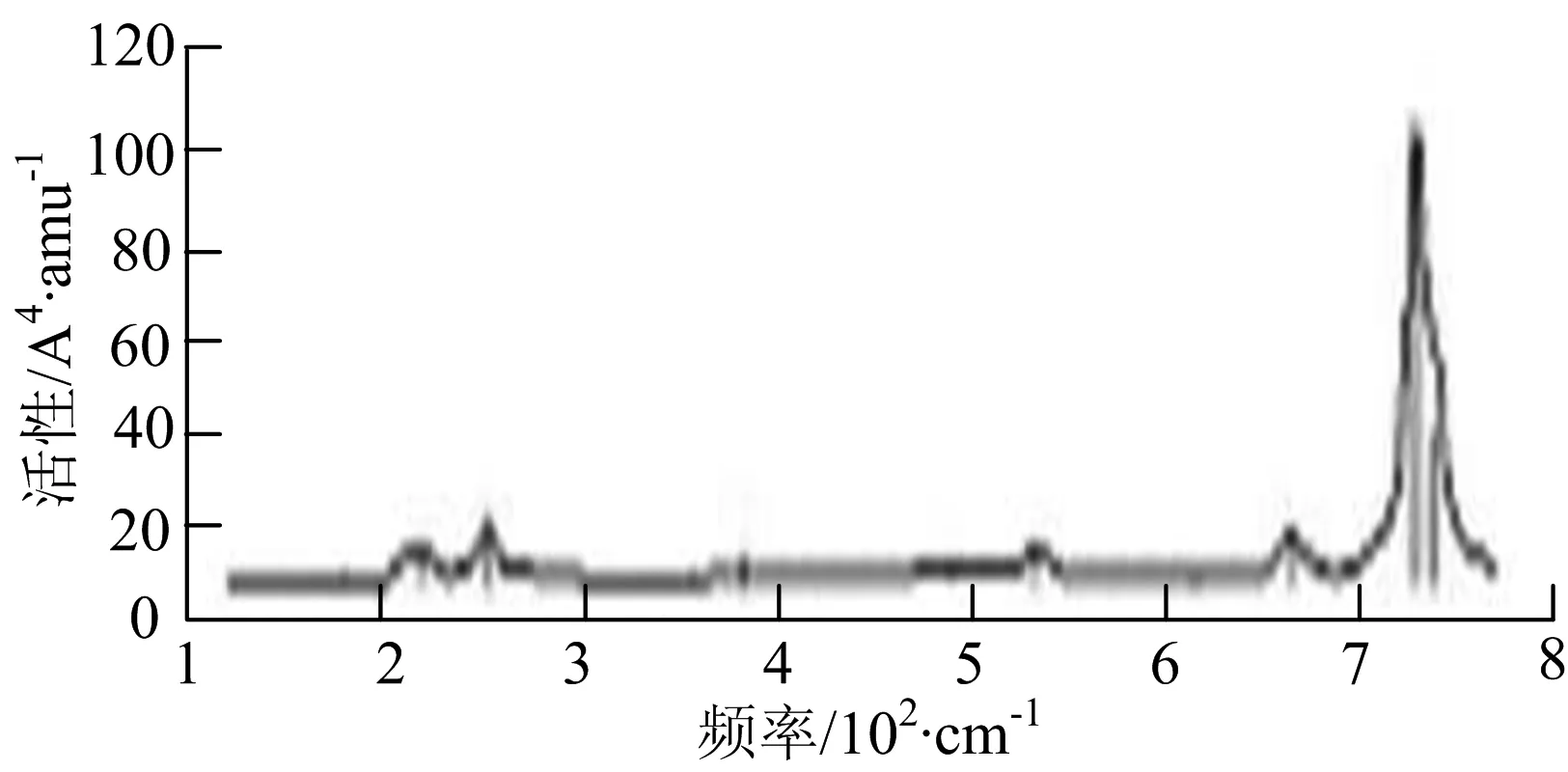
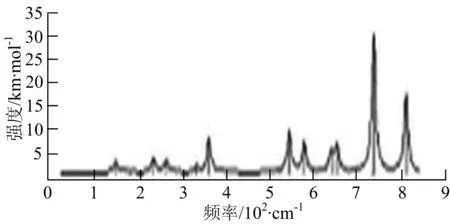
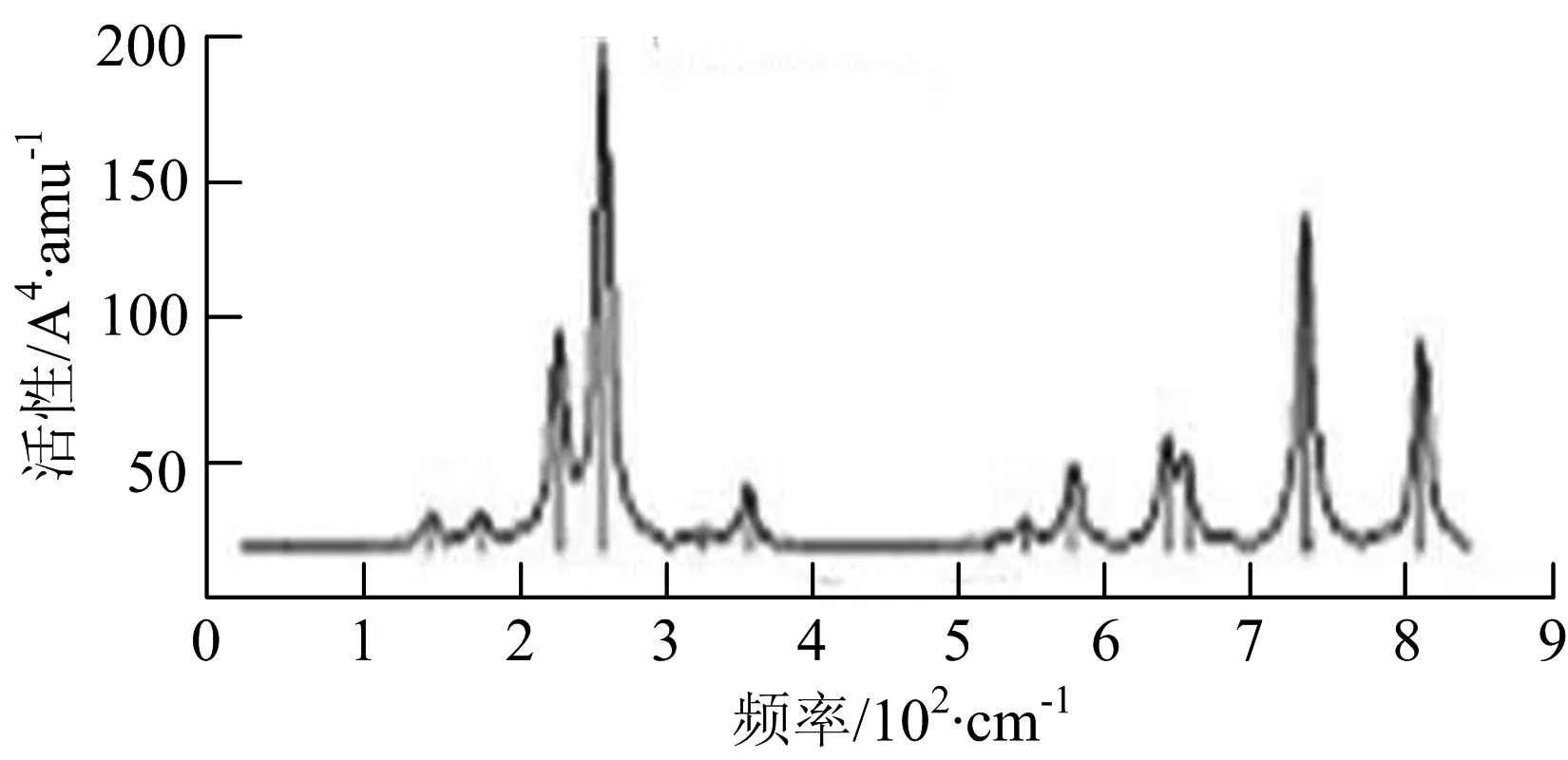
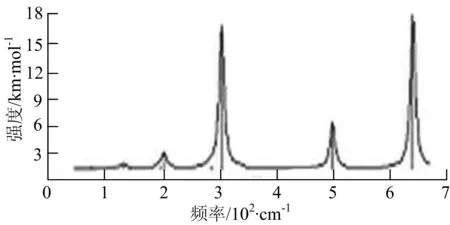
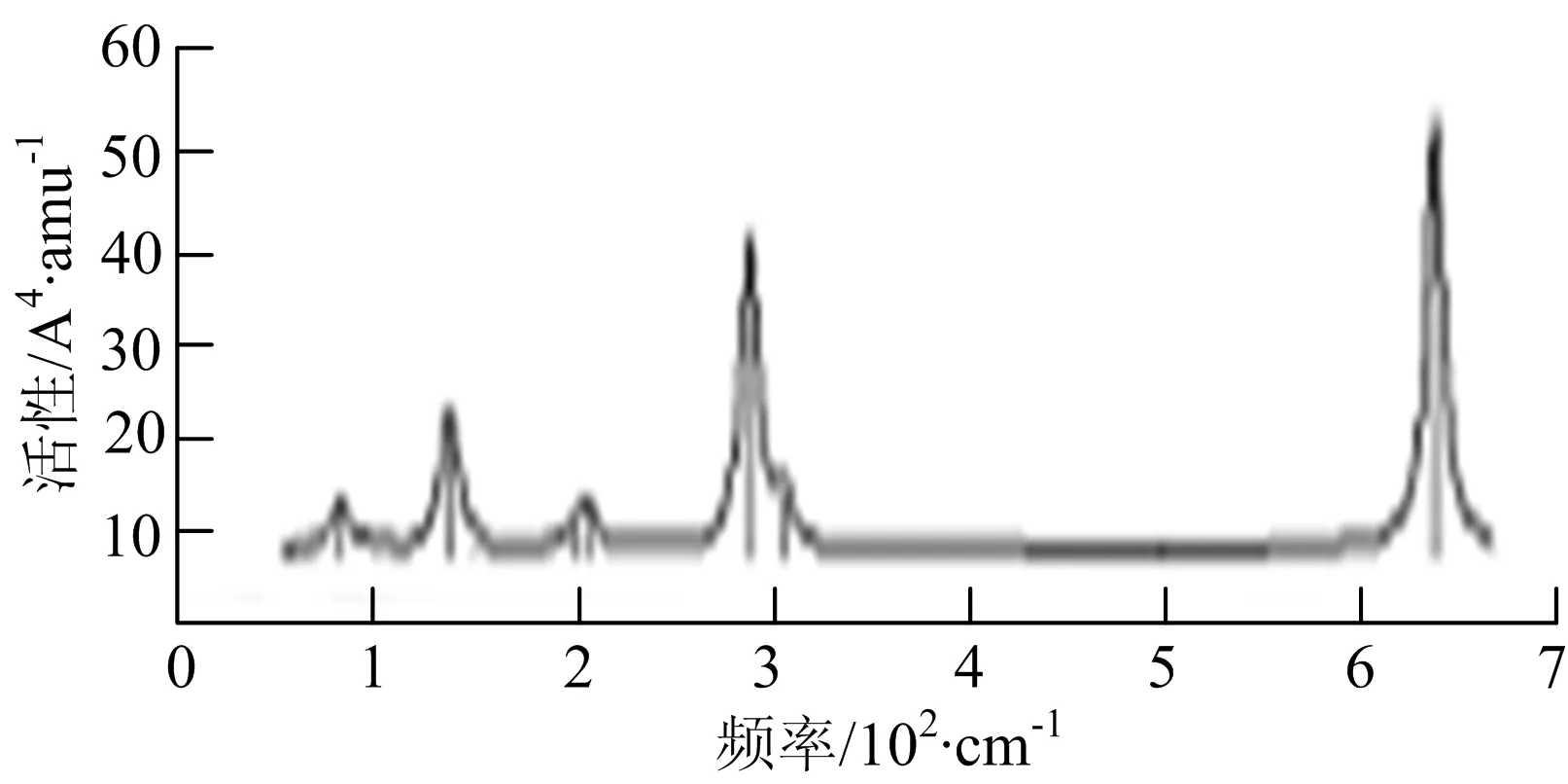
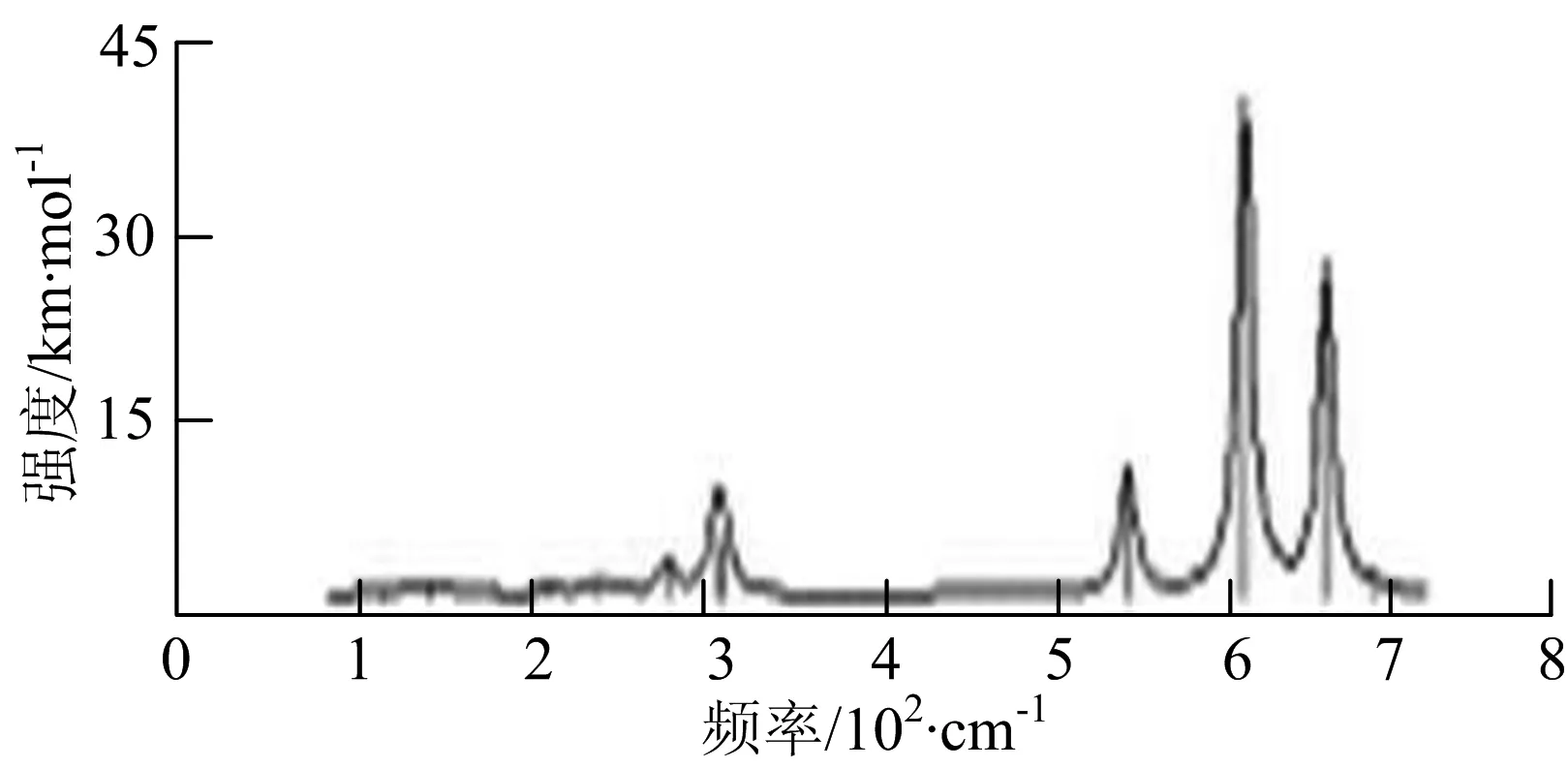
图2给出了WmBn(m+n≤7)团簇基态结构的红外(IR)光谱图和拉曼(Raman)光谱图.通过GaussView来判定各团簇光谱峰值所对应频率的振动方式的归属情况.
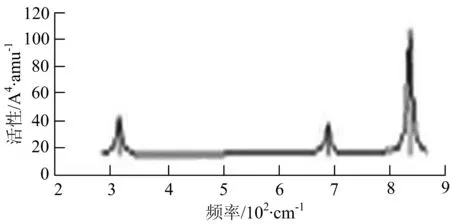
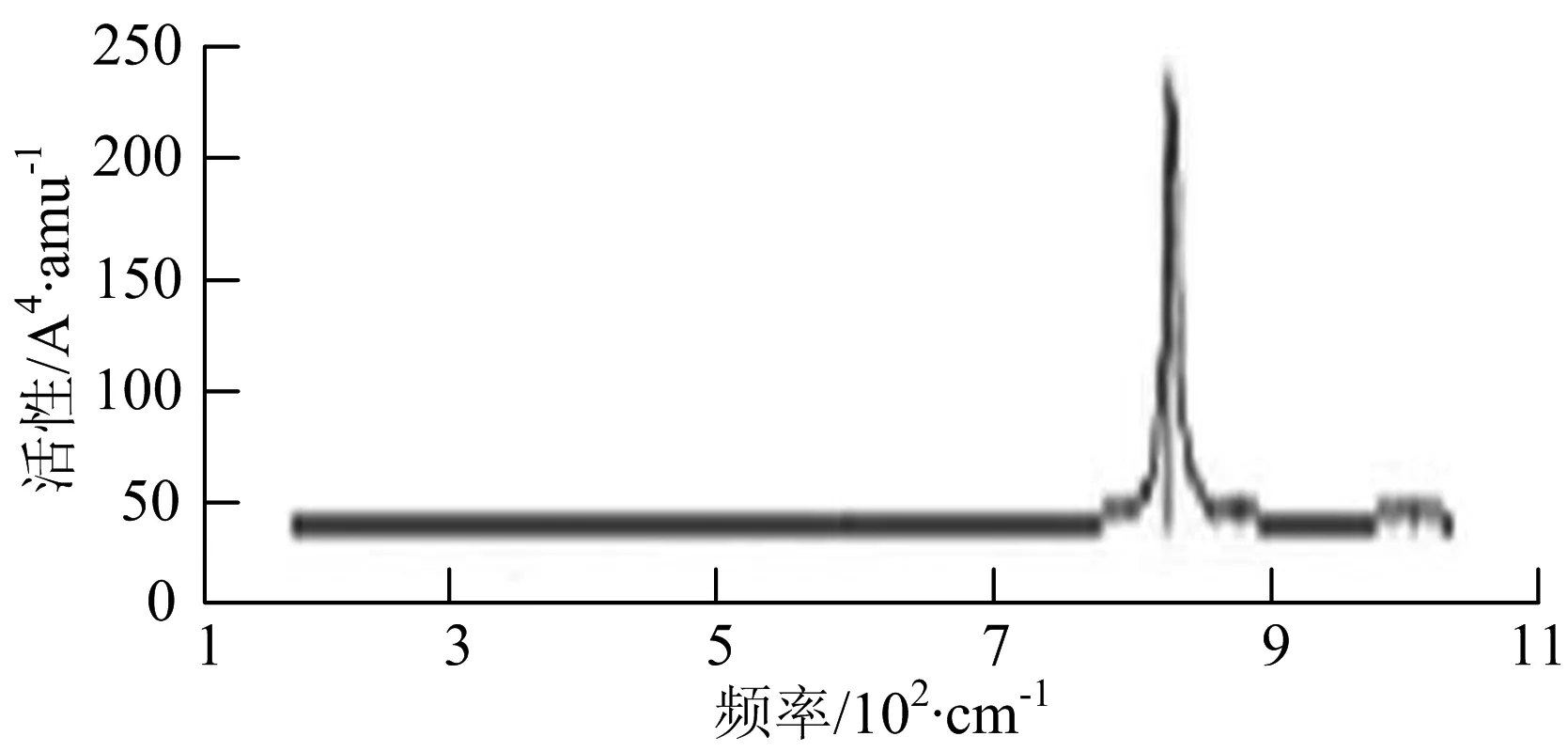
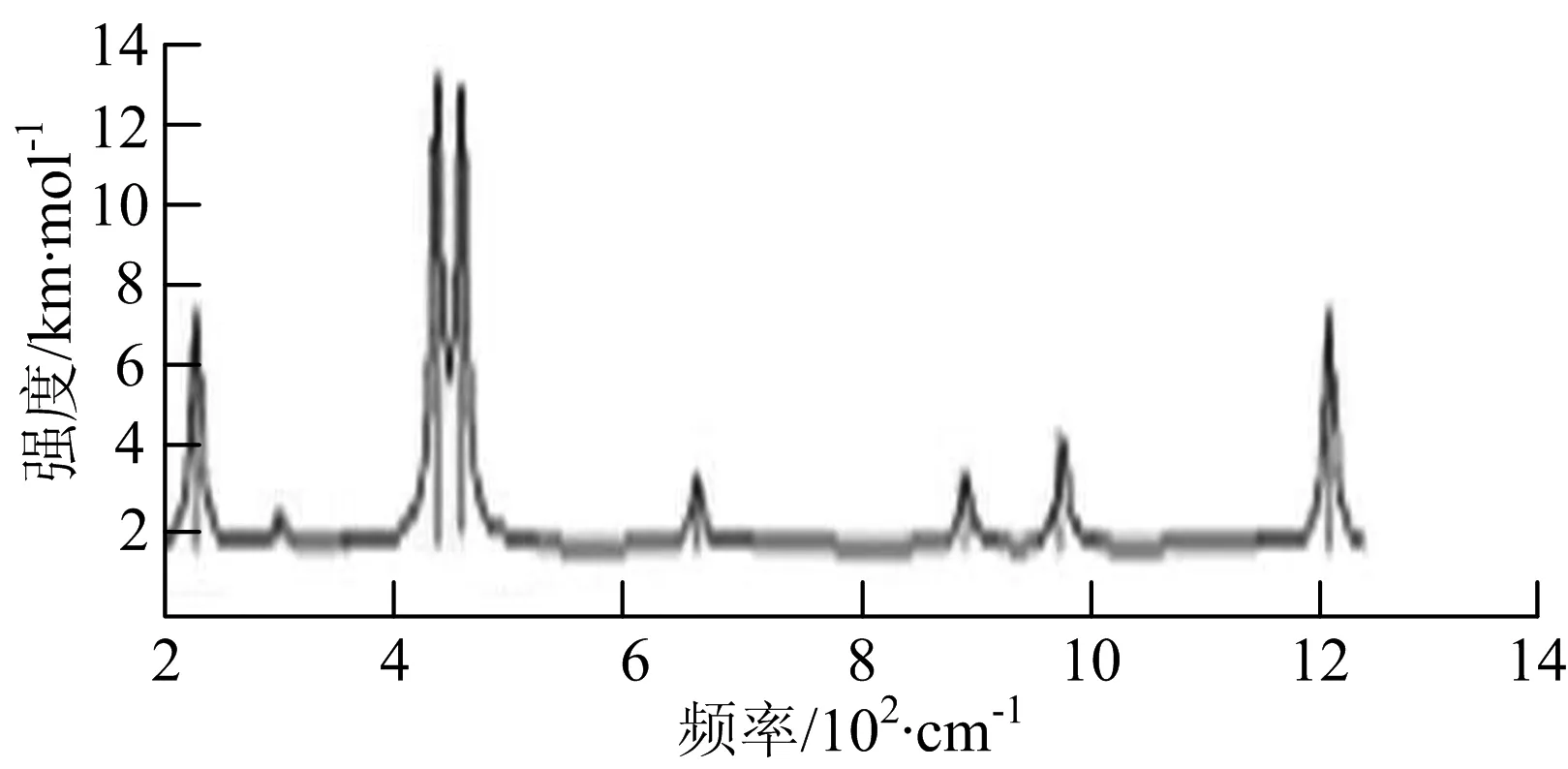
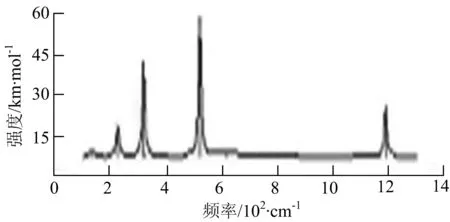
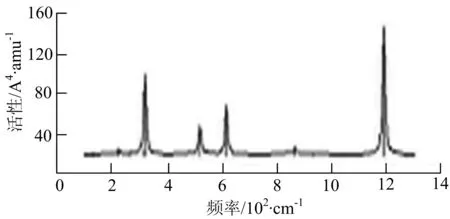
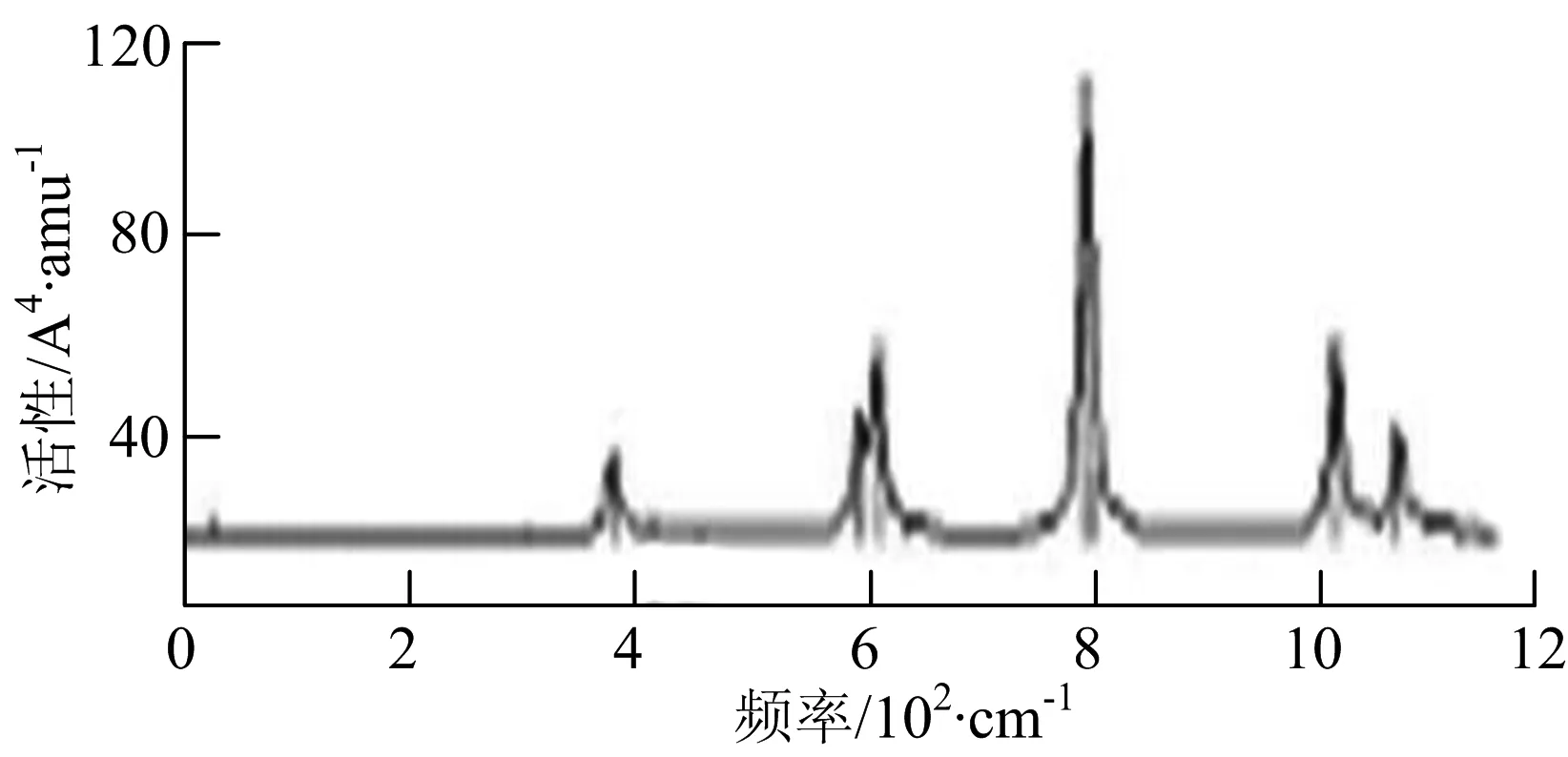
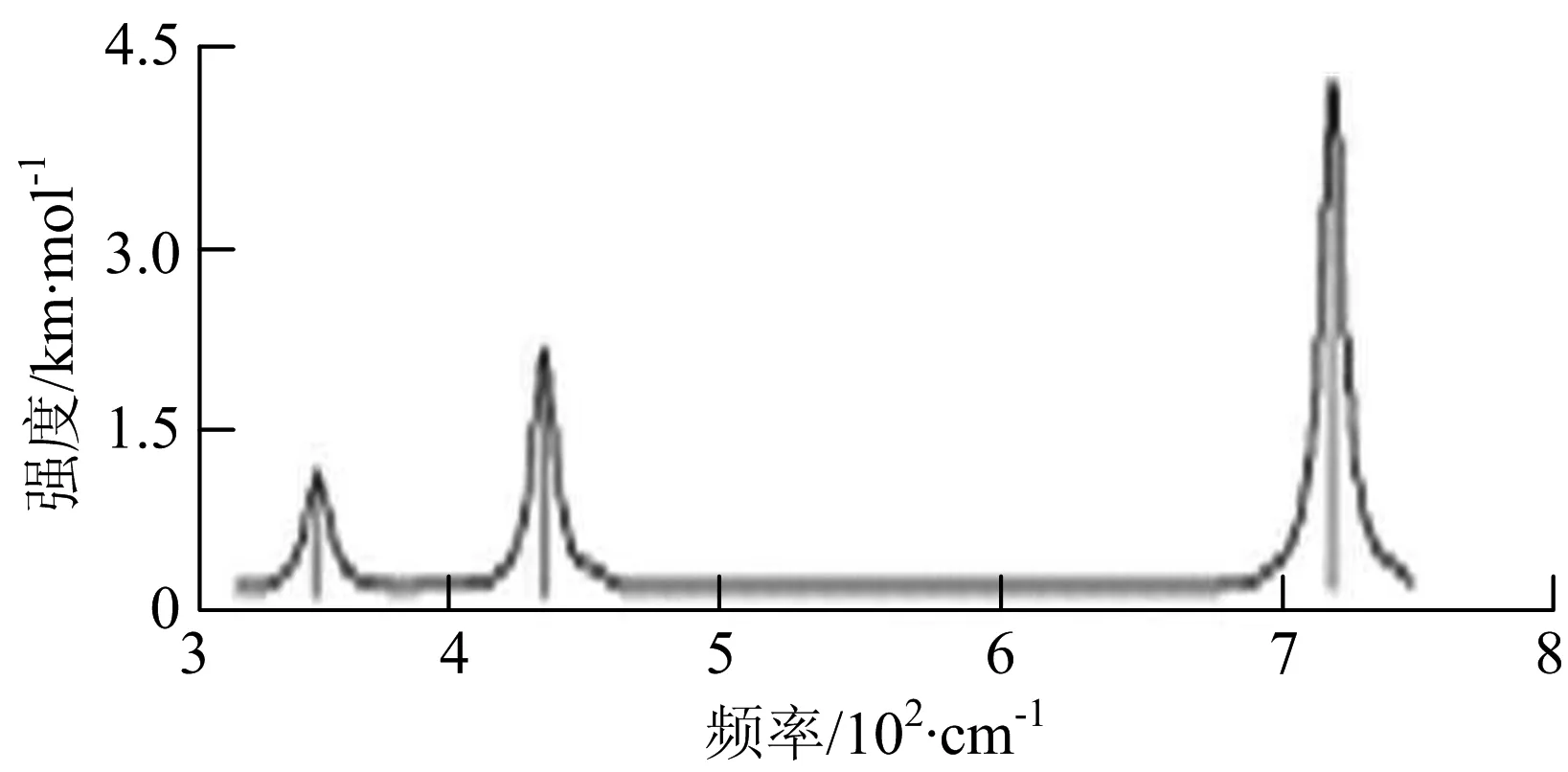
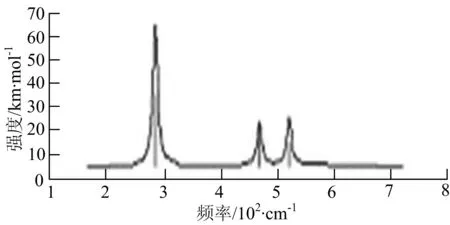
由图2可以看出,WB2团簇红外光谱只有1个振动峰位于频率695 cm-1处,该处的振动模式为2个B原子沿W-B键的方向做不对称伸缩振动;拉曼光谱中有3个振动峰,其最强振动峰位于频率847 cm-1处,该处的振动模式为2个B原子沿W-B键的方向做对称伸缩振动.WB3团簇红外光谱有4个振动峰,其最强振动峰所对应频率1 010 cm-1处,振动方式为3个B原子沿着B-B键方向往返伸缩振动,在振动过程中化学键的键长发生变化,次强峰位于频率877cm-1处,该处的振动模式为2个B原子沿W-B键的方向做不对称伸缩振动,从而引起化学键键长的变化;拉曼光谱中有只有1个振动峰,位于频率827 cm-1处,其振动模式为W原子和其中1个B原子固定不动,其他2个B原子做对称伸缩振动;WB3团簇的IR和Raman主峰都在高频段,表明在高频段该团簇红外和拉曼活性都很强.WB4团簇红外光谱有多个振动峰,其中最强振动峰位于频率443 cm-1处,其振动模式为B原子做面内弯曲振动;拉曼光谱和红外光谱一样,几乎整个频段上都有峰值分散出现,其最强振动峰位于频率995 cm-1处,其振动模式为不对称伸缩振动;次强峰位于波数672 cm-1处,此时2个B原子做不对称伸缩振动,而引起的结构变形以及化学键键长的改变.WB5团簇红外光谱有4个振动峰,最强峰位于频率529 cm-1处,其振动模式为平面剪式振动;拉曼光谱中有多个振动峰,最强振动峰位于频率1 205 cm-1处,振动模式为平面对称伸缩振动.
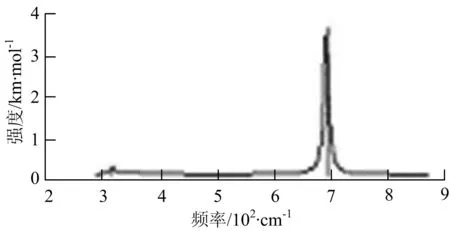
a)WB2
b) WB3
c) WB4
d) WB5
e) WB6
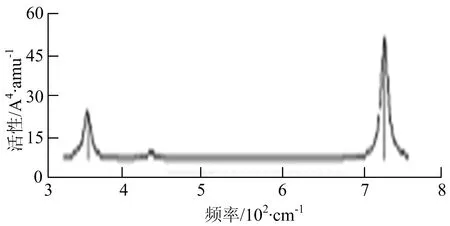
f) W2B
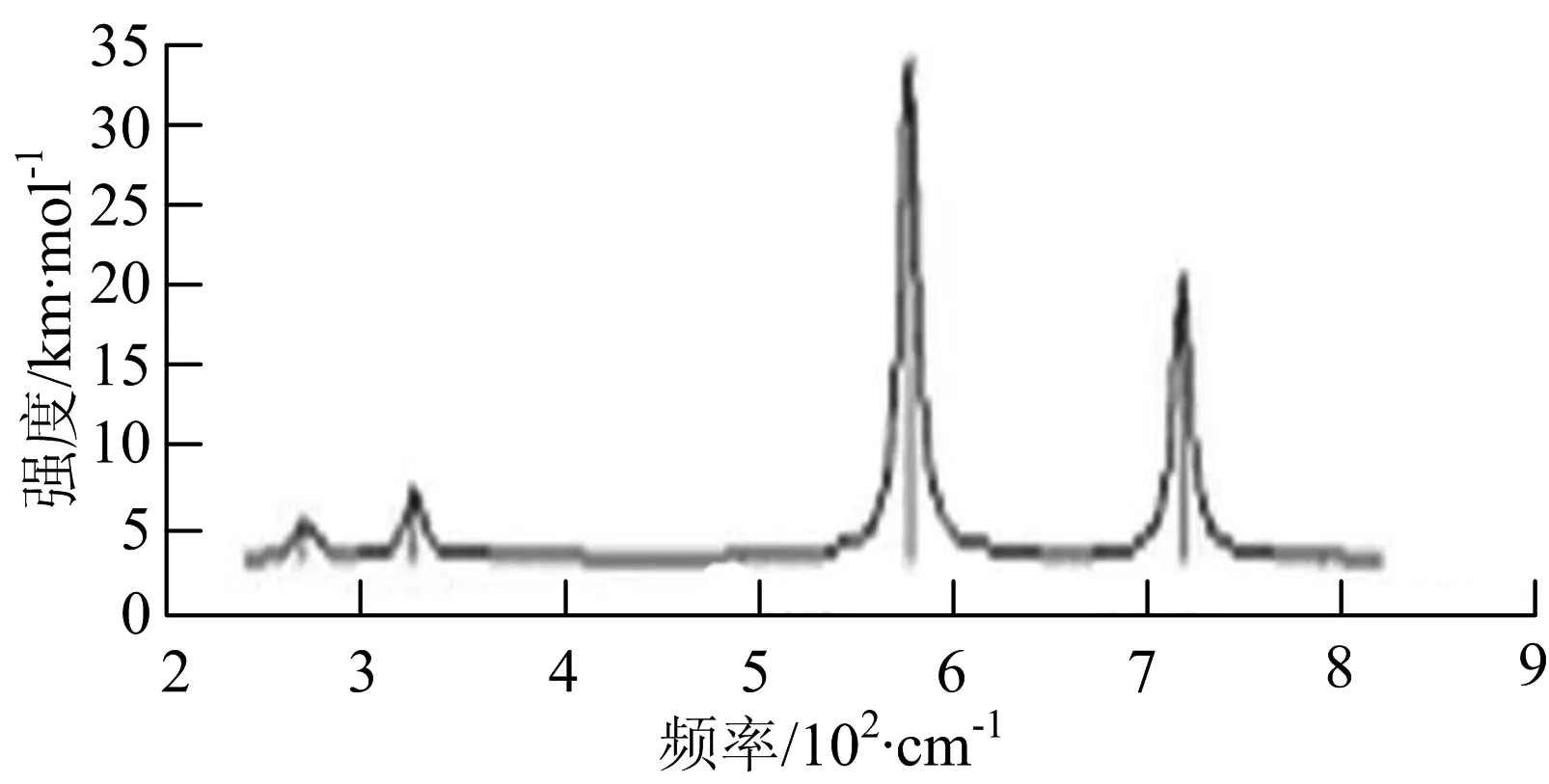
g) W2B2
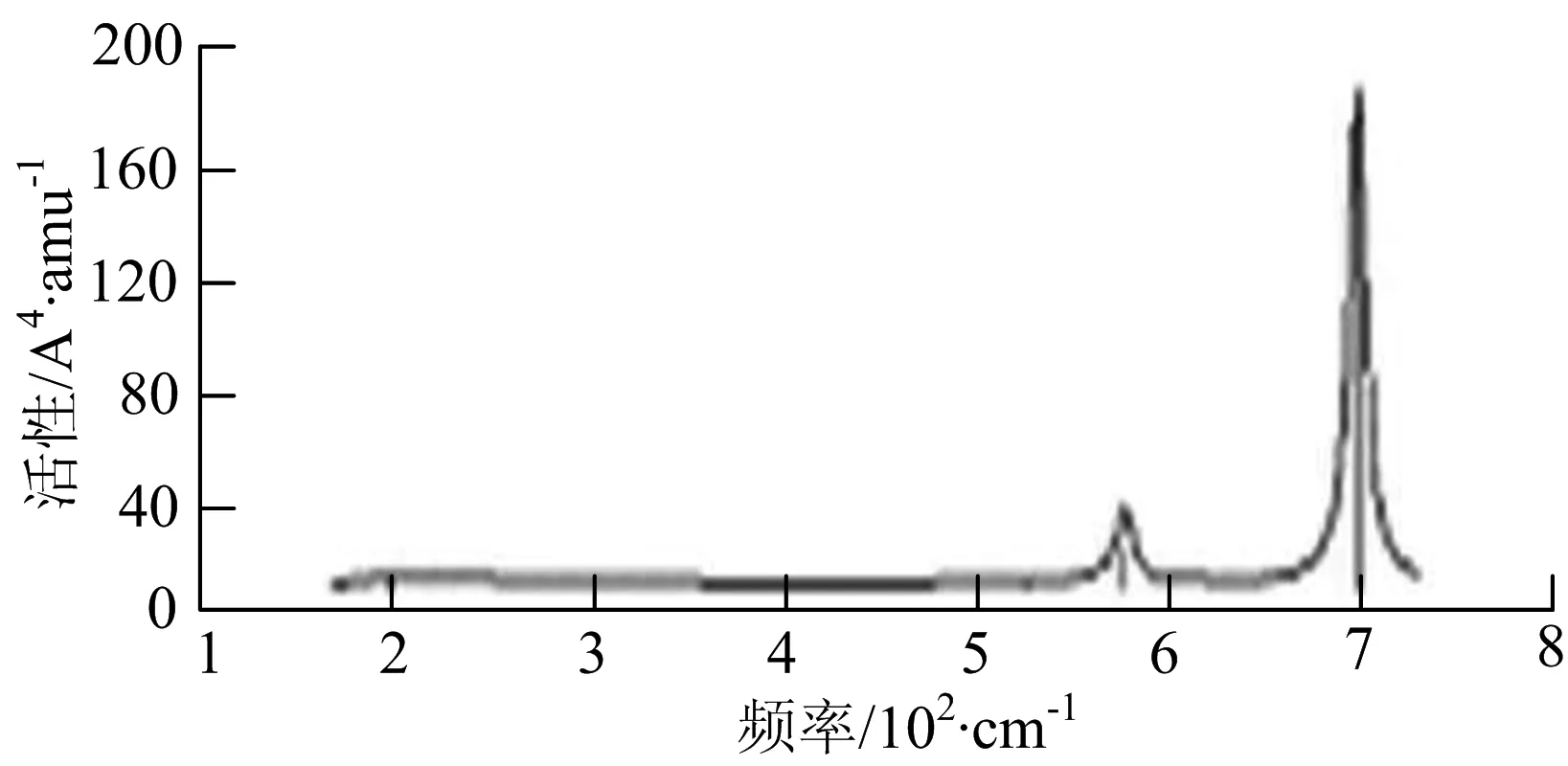
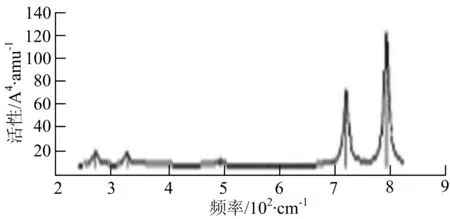
h) W2B3
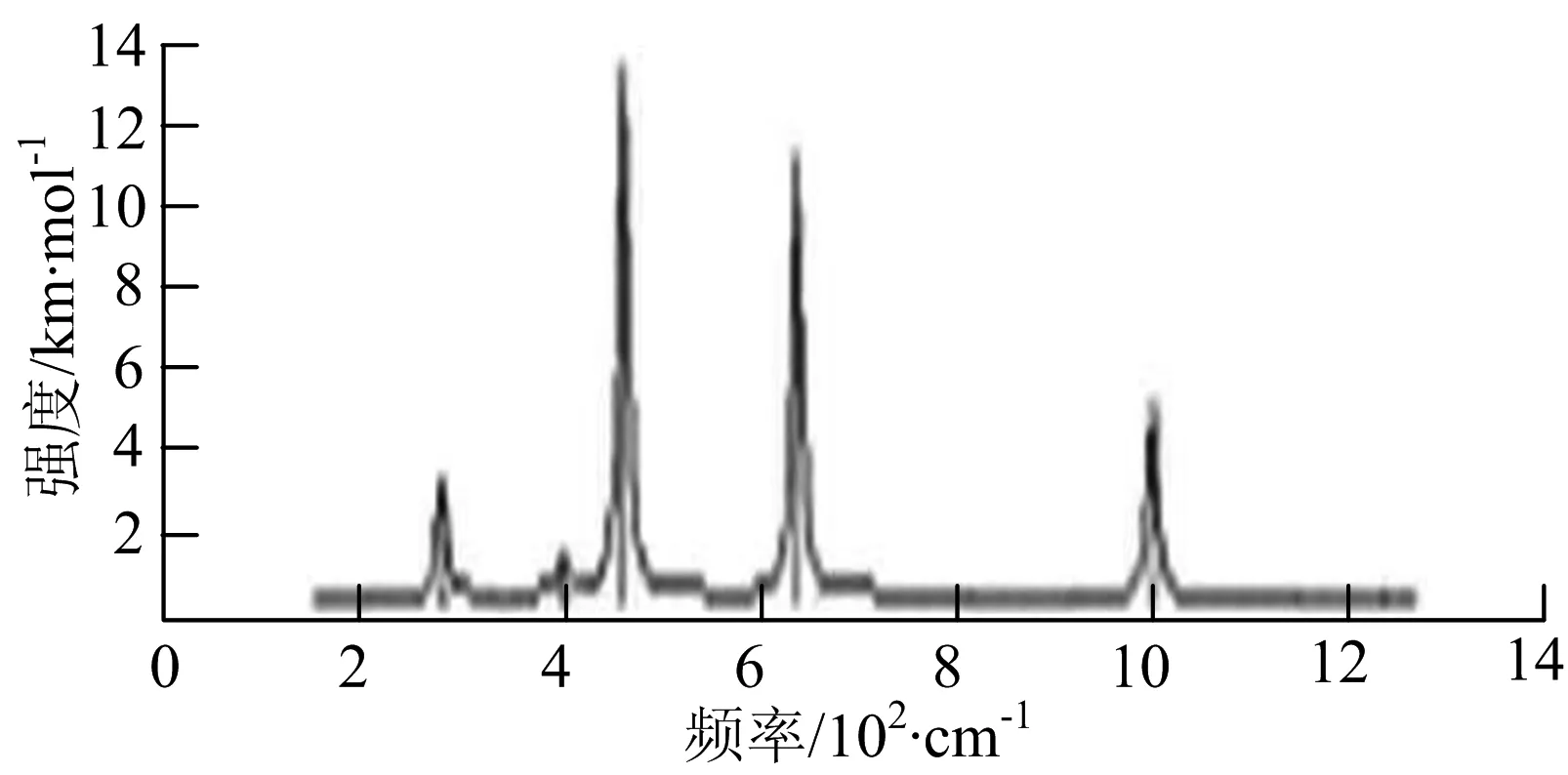
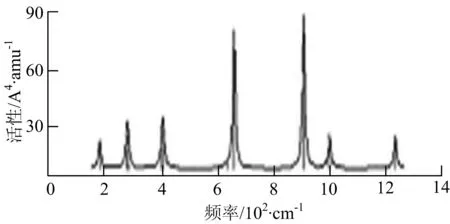
i) W2B4
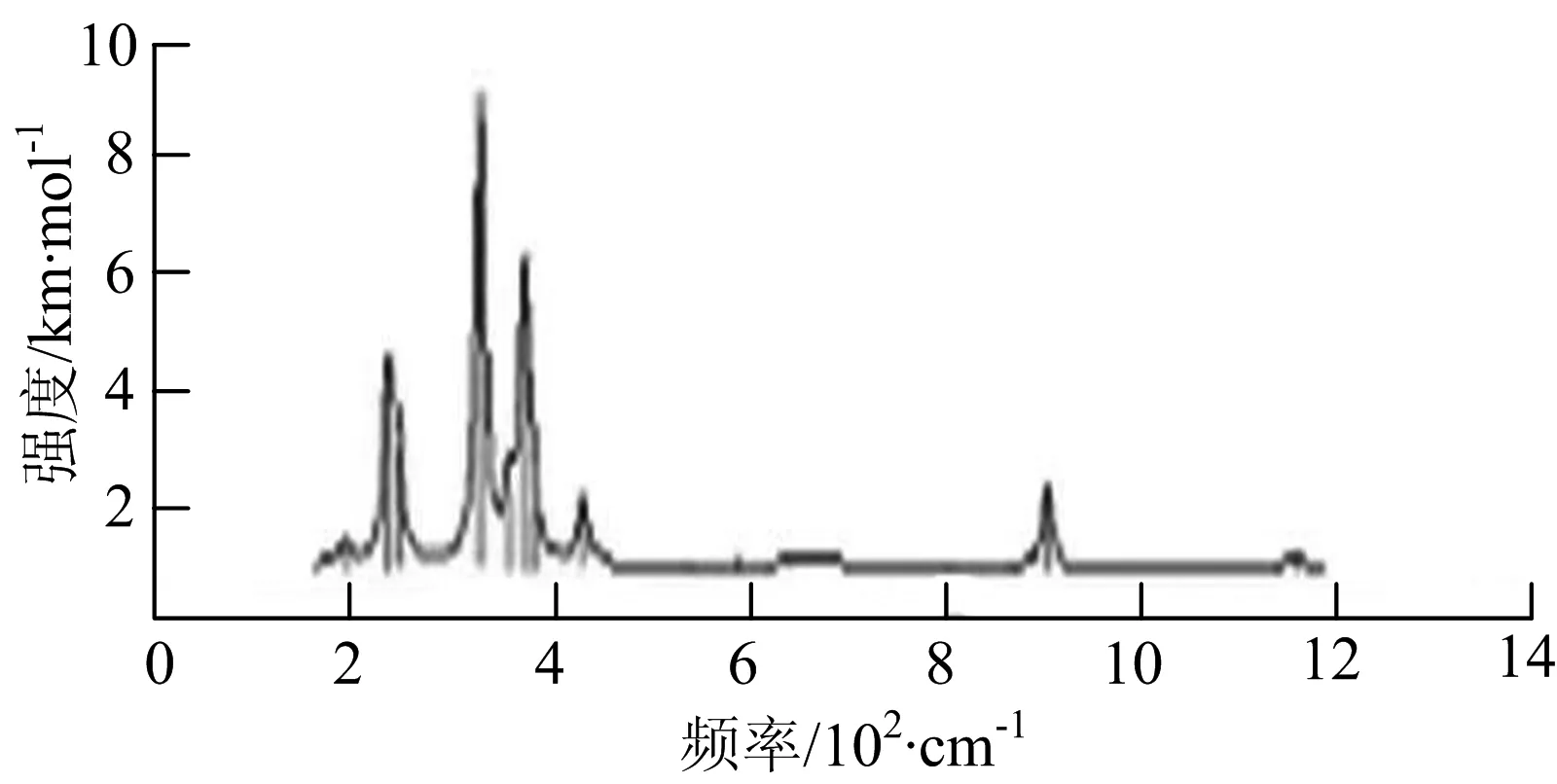
j) W2B5
k) W3B
l) W3B2
m) W3B3
n) W3B4
o) W4B
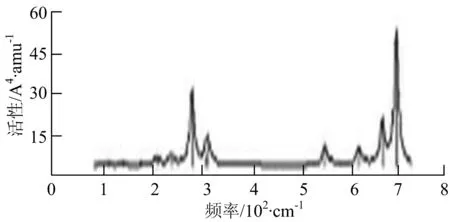
p) W4B2
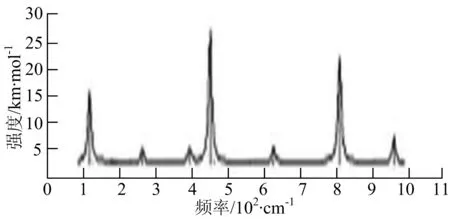
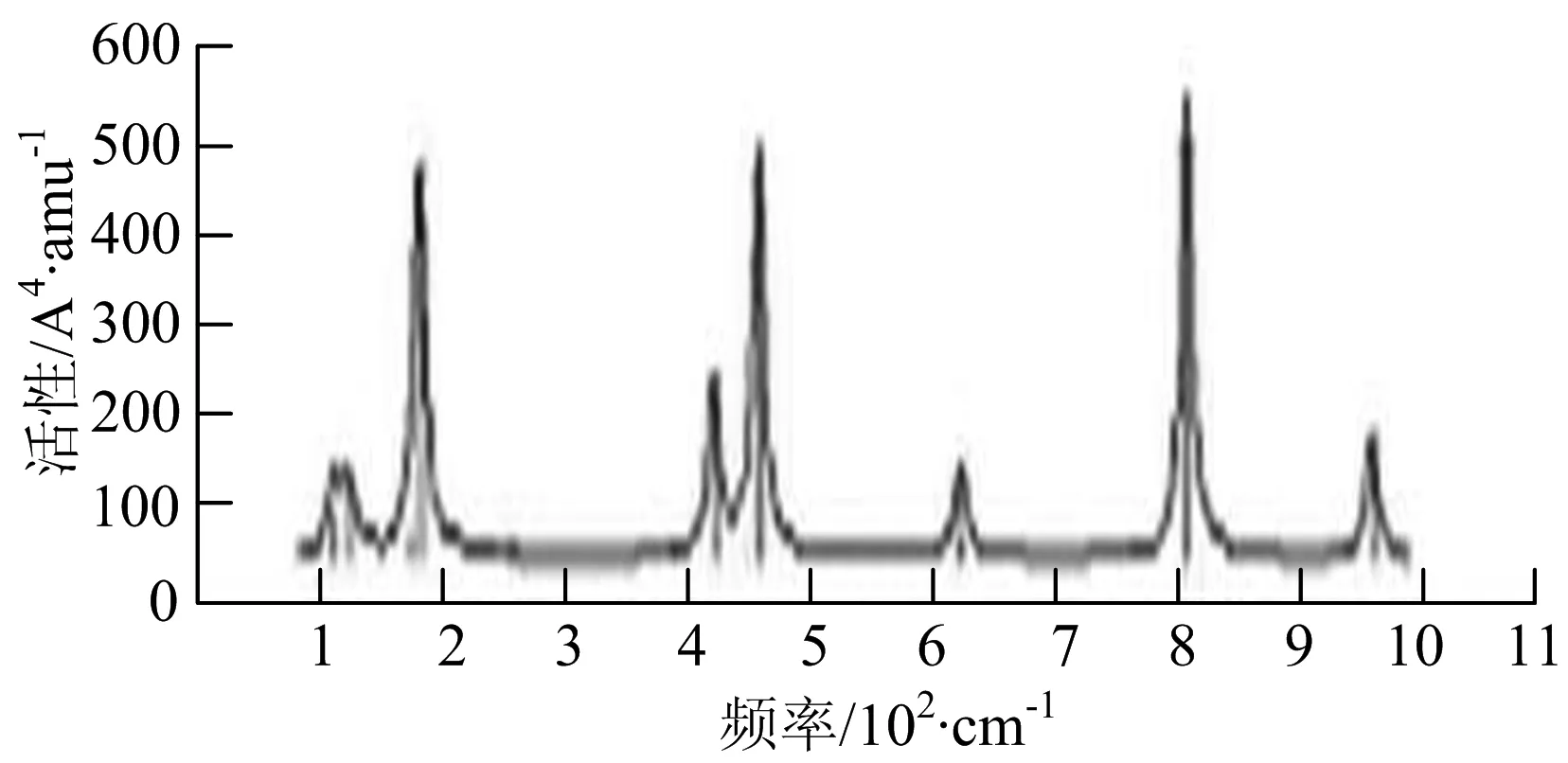
q) W4B3
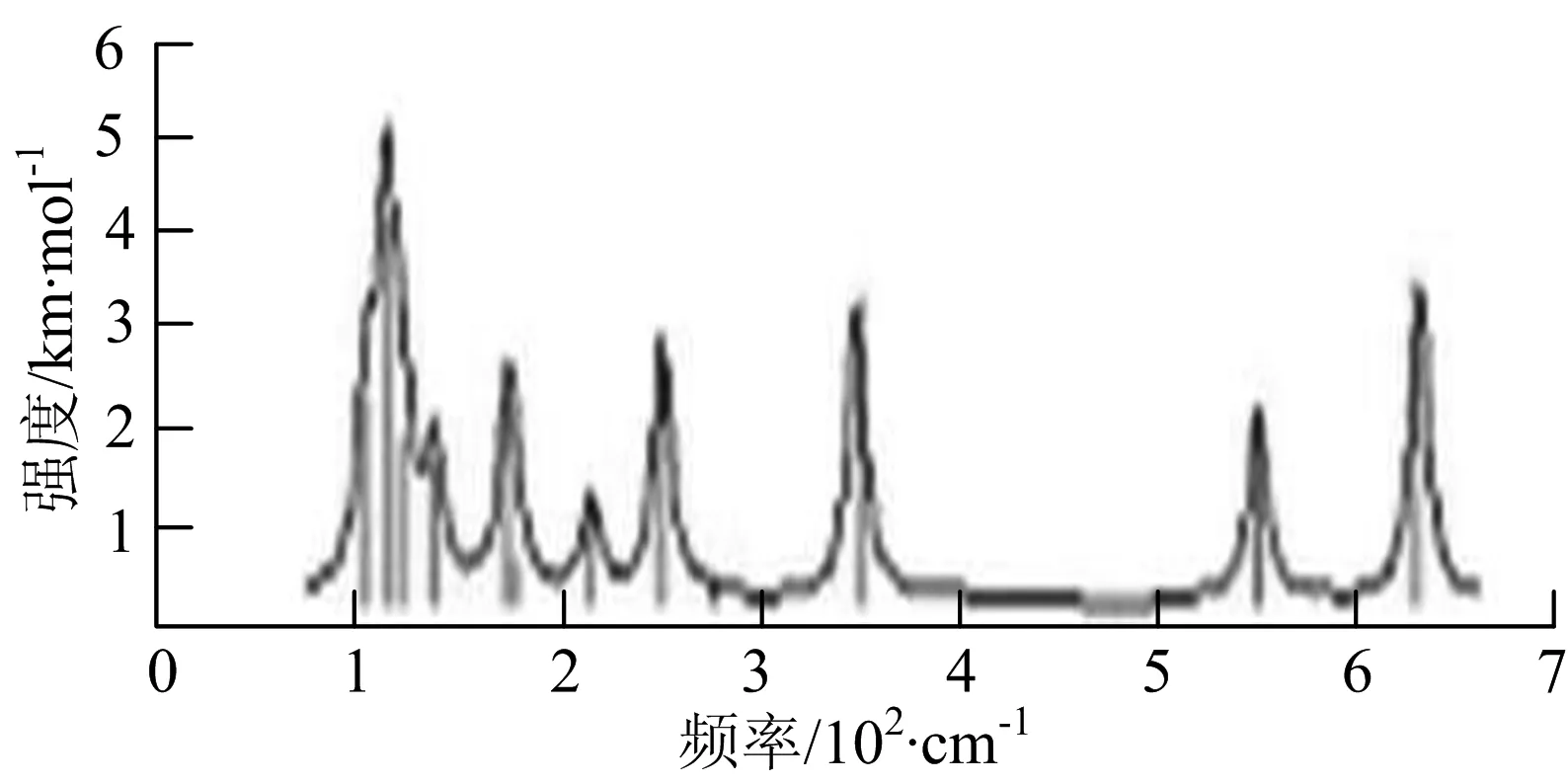
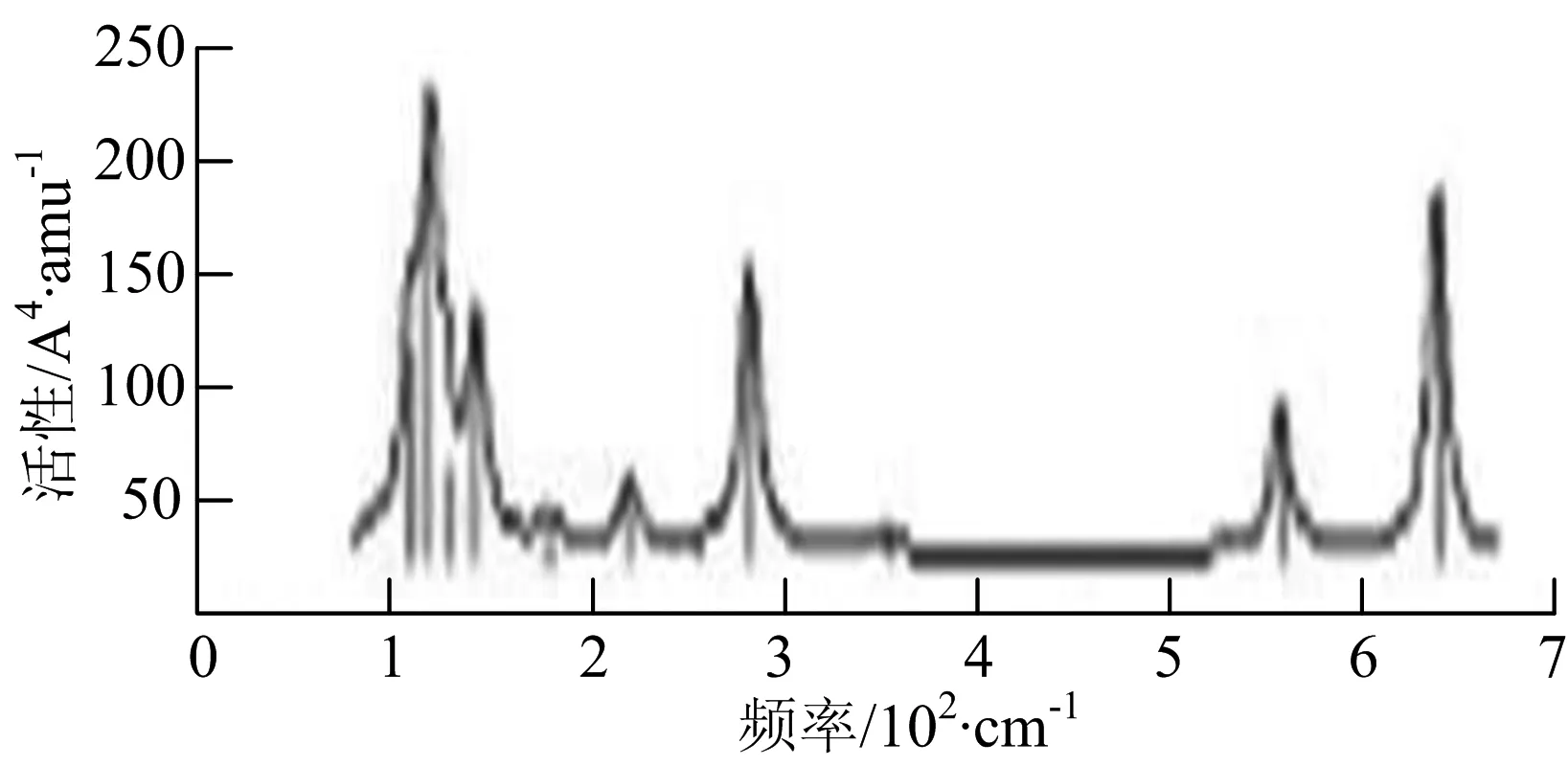
r) W5B
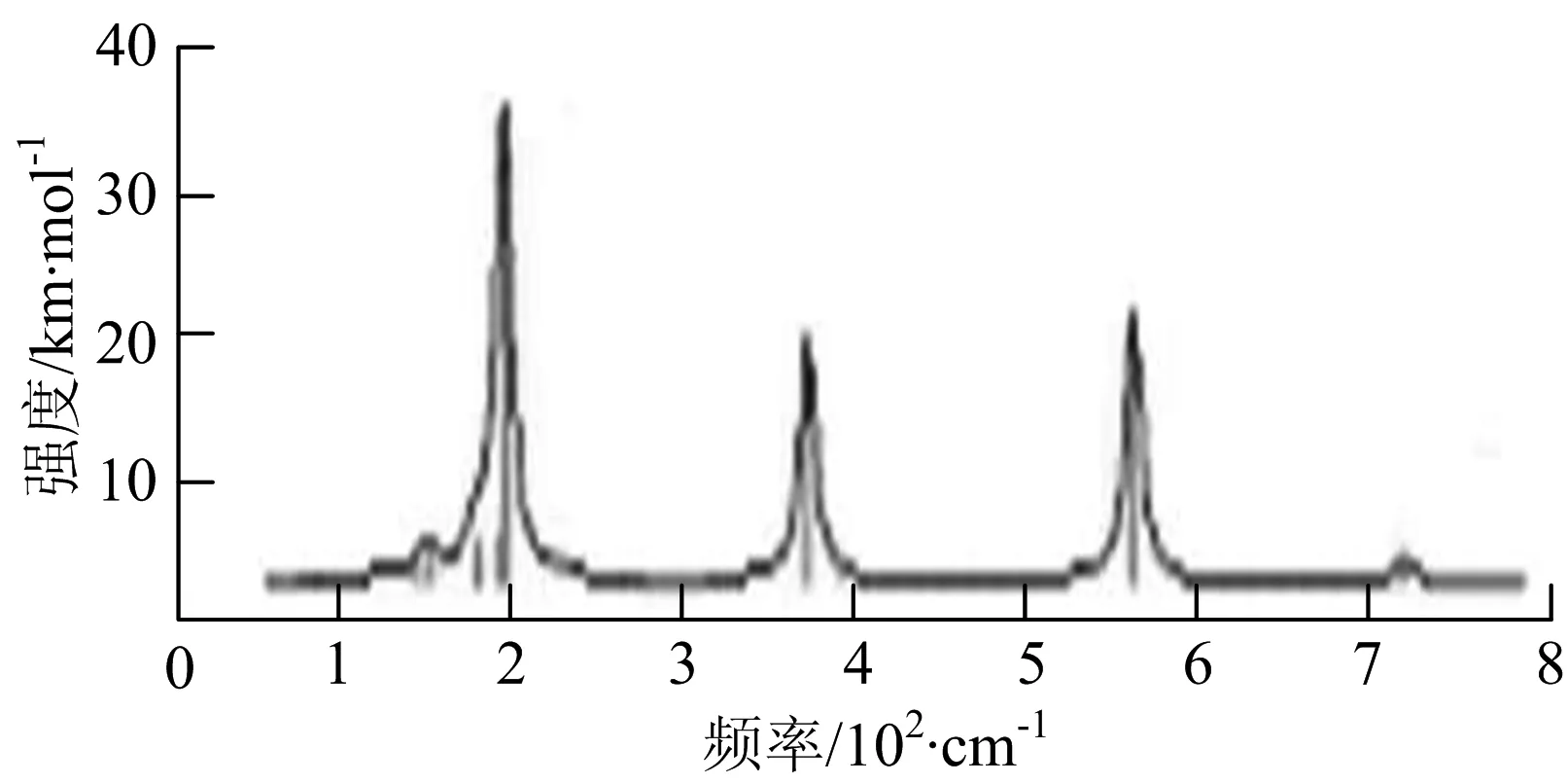
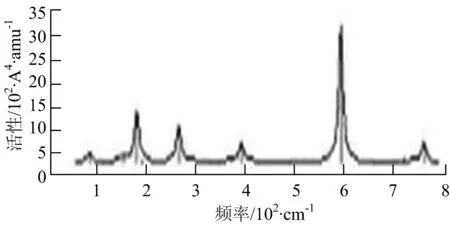
s) W5B2
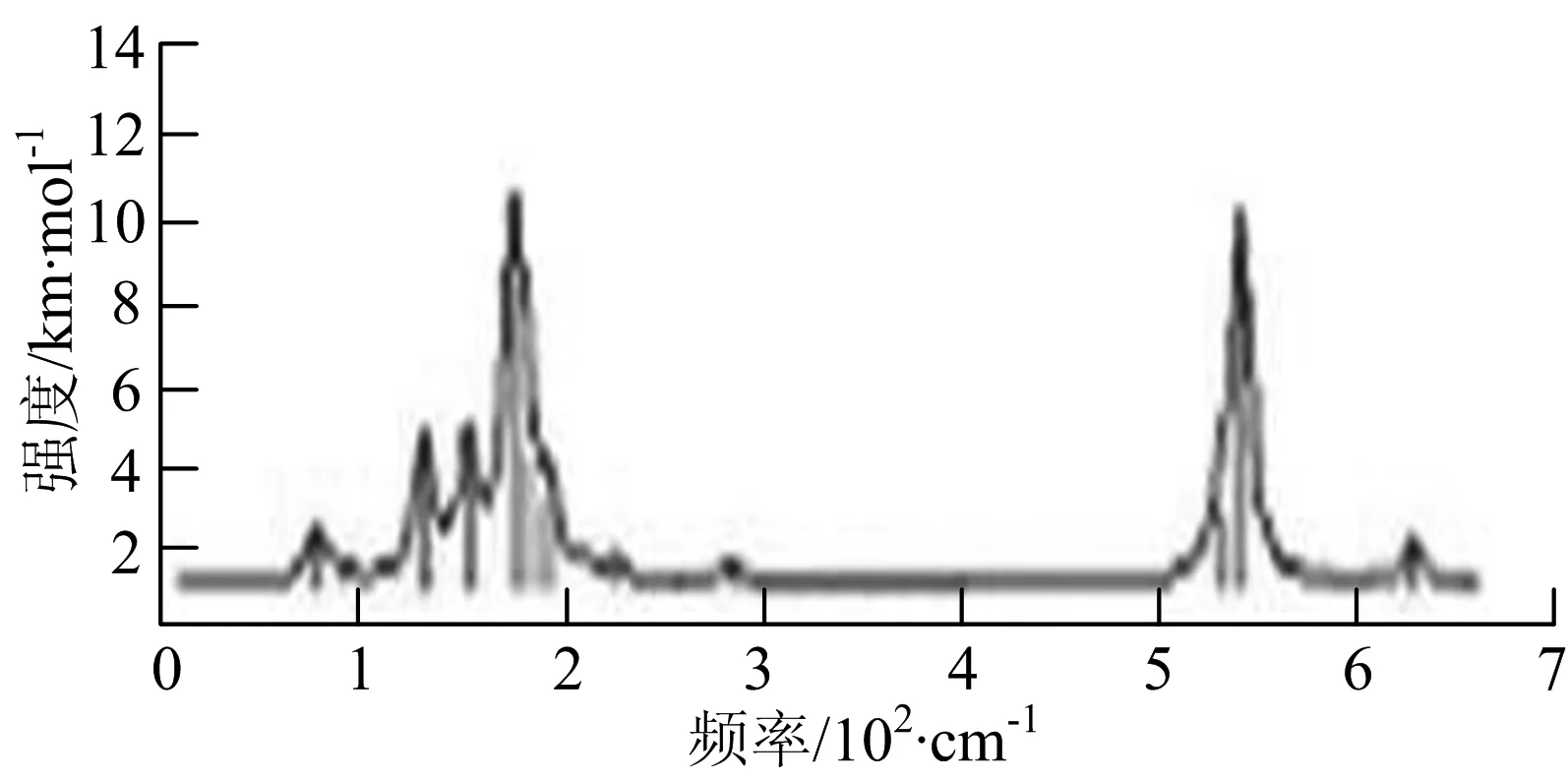
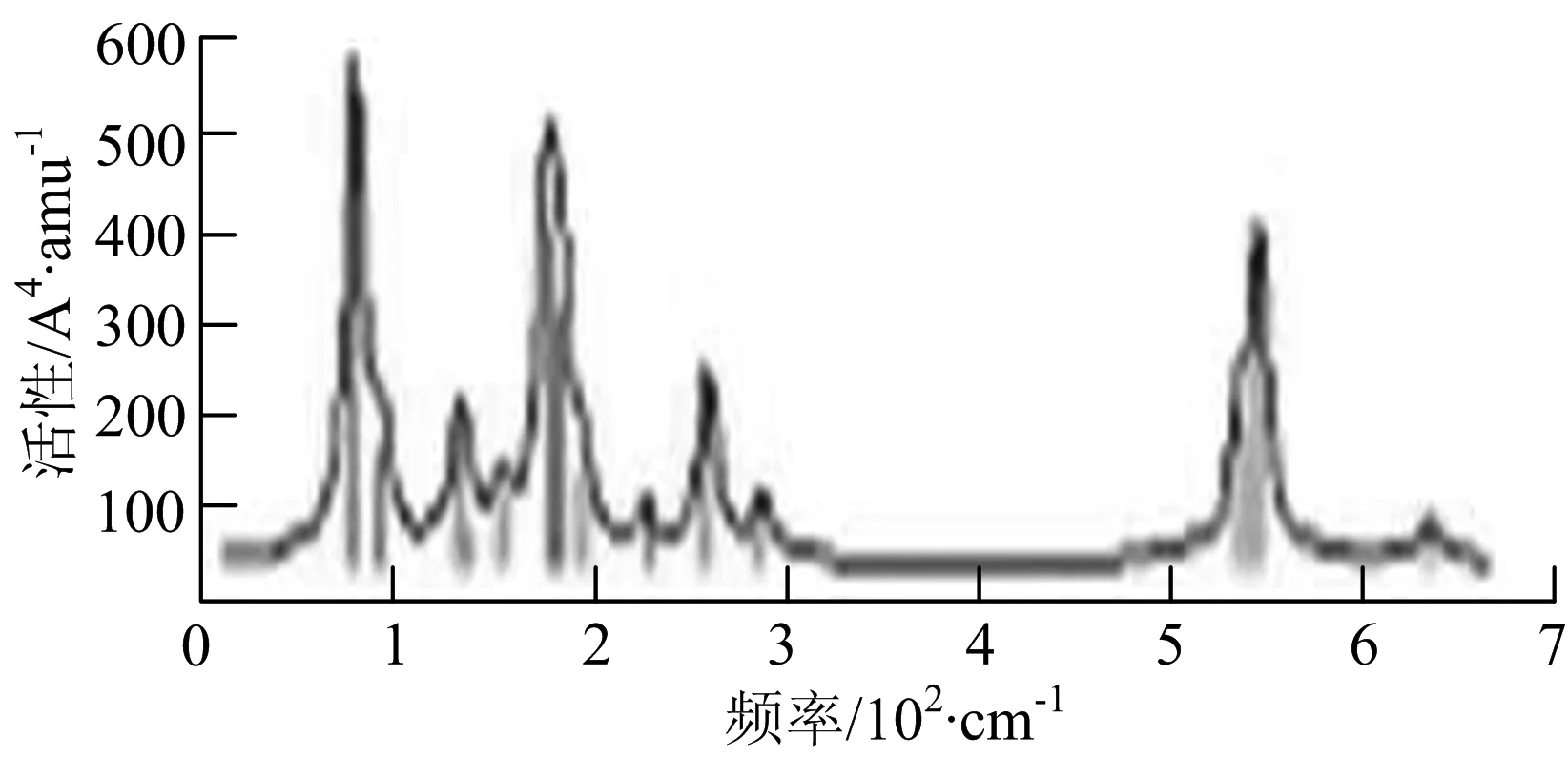
t) W6B
WB6团簇红外光谱在1 000 ~1 200 cm-1段内有连续多个峰值,说明在这个频段内该团簇红外活性很好,且主峰值相差较小,最强振动峰位于频率1 112 cm-1处,振动模式为位于几何中心的B原子做不对称伸缩振动;拉曼光谱中有多个振动峰,最强振动峰位于频率805cm-1处,振动模式为平面剪式振动.
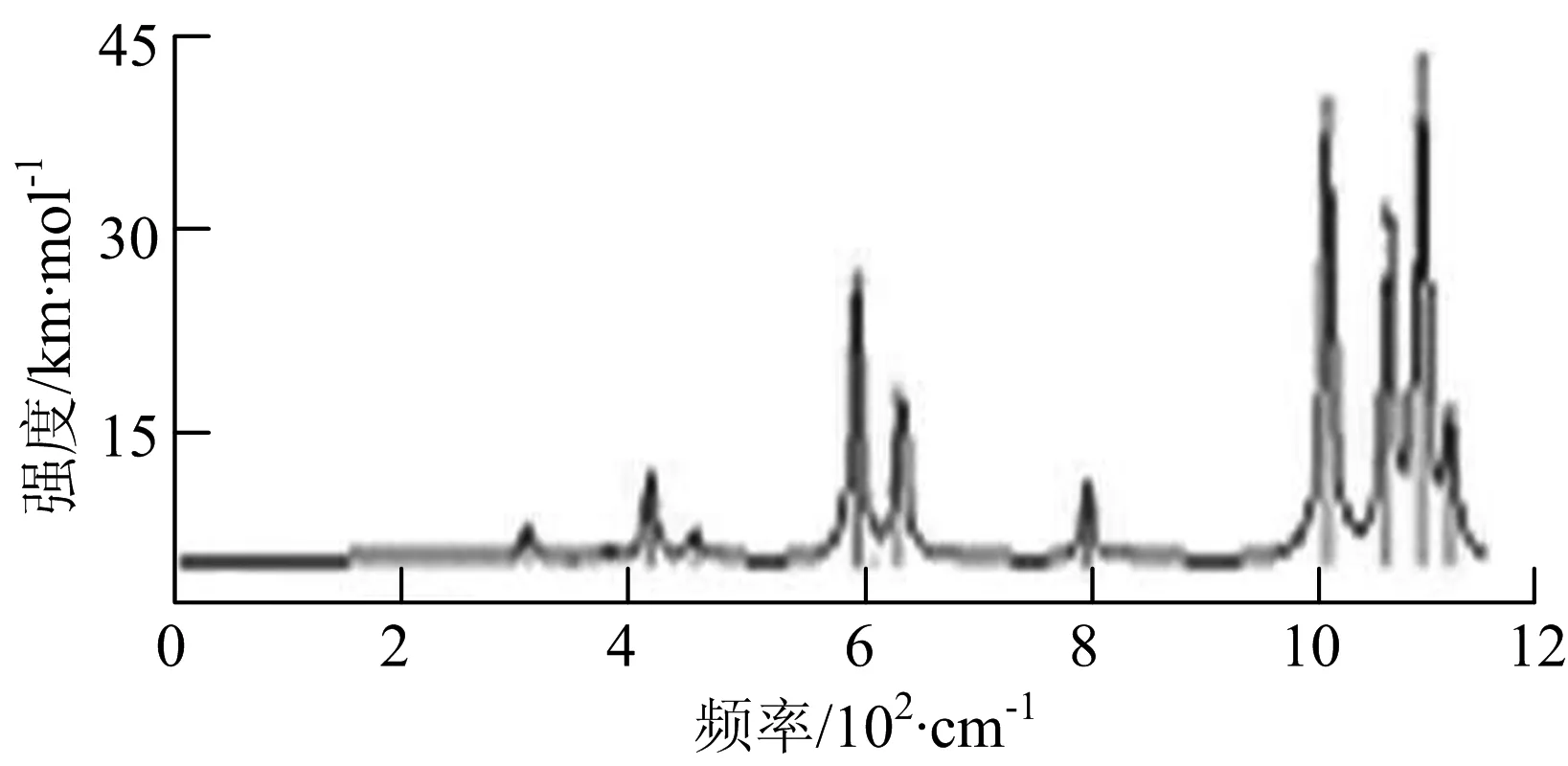
W2B团簇红外光谱有3个振动峰,最强峰的振动模式是B原子的对称伸缩振动,位于频率727 cm-1处;拉曼光谱有3个振动峰,最小峰强度小的几乎看不出来,最强峰的振动模式为平面对称伸缩振动,位于727 cm-1处.W2B2团簇红外光谱有4个振动峰,最强振动峰位于频率为584 cm-1处,振动模式为2个B原子在各自的平面内做摇摆振动;拉曼光谱中有多个振动峰,最强峰位于频率为797 cm-1处,振动模式为2个B原子分别沿W-B键做对称伸缩振动.W2B3团簇红外光谱有3个振动峰,在频率288 cm-1处的是最强峰,振动模式为下方2个B原子的上下摇摆振动; 拉曼光谱中只有2个振动峰,最强峰位于频率为700 cm-1处,振动模式为呼吸振动.W2B4团簇红外光谱有多个振动峰,最强振动峰位于467 cm-1处,振动模式为位于两翼的B原子做面外摇摆振动;拉曼光谱的振动峰几乎分布在整个频段上,最强峰位于913 cm-1处,振动模式为各个B原子在同一平面内做剪式振动.W2B5团簇红外光谱在200~450 cm-1段内有多个连续的峰值,但强度都很小,其中最强振动峰峰位于339 cm-1出,振动模式为面外弯曲振动;拉曼光谱的峰值几乎分散整个频段,在1 179 cm-1处的振动最强,振动模式为沿B-B键方向的对称伸缩振动.
W3B团簇的红外光谱在258 cm-1处出现了最强谱峰,是由于B原子做面外摇摆振动产生的;拉曼光谱和红外光谱类似,分布在相对较高和较低的频段,在718 cm-1处出现了最大谱峰,是由于W原子之间的相互伸缩振动而产生的.W3B2团簇红外光谱和拉曼光谱都有多个振动峰,红外光谱最强振动峰位于545 cm-1处,振动模式为面内弯曲振动;拉曼光谱最强振动峰位于545 cm-1处,同红外光谱相同,振动模式为面内弯曲振动.W3B2团簇与WB4团簇的分布情况类似,IR和Raman的峰值比较分散,几乎整个频段上都有.W3B3团簇红外光谱有多个振动峰,分布在整个频段上,最强振动峰位于621 cm-1处,位于左右下方的2个B原子做前后面外摇摆振动;拉曼光谱虽然有多个振动峰,但是只有一个显著的峰,位于735 cm-1处,该振动模式为3个B原子沿W-B键做对称伸缩振动.W3B4团簇红外光谱分布在整个频段上,最强振动峰位于739 cm-1处,振动模式为扭曲振动;拉曼光谱也分散在整个频段,最强振动峰位于269 cm-1处,振动模式为剪式振动.
W4B团簇红外光谱有多个较强振动峰,最强振动峰位于646 cm-1处,振动模式为B原子的伸缩振动;拉曼光谱的最强振动峰位于646 cm-1处,振动模式为B原子的伸缩振动.W4B2团簇的红外光谱和拉曼光谱峰都比较多,而且都是分段集中;红外光谱最强振动峰位于627 cm-1处,振动模式为2个B原子的前后不对称摇摆振动;拉曼光谱的最强振动峰位于703 cm-1处,振动模式为2个B原子的前后对称伸缩振动.W4B3团簇红外光谱和拉曼光谱都有多个振动峰,而且都比较分散;红外光谱最强振动峰位于456 cm-1处,振动模式为位于顶端的B原子的摇摆振动;拉曼光谱最强振动峰位位于822 cm-1处,振动模式为3个B原子沿B-B键做伸缩振动.
W5B团簇红外光谱在100~260 cm-1频段上有多个连续的峰值,最强振动峰位于118 cm-1处,振动模式为W-W键和W-B键的伸缩振动;拉曼光谱有多个振动峰,最强振动峰位于118 cm-1处,振动模式为W-W键和W-B键的伸缩振动.W5B2团簇红外光谱只有3个振动峰,193 cm-1处的最强振动峰振动模式为伸缩振动;拉曼光谱有多个振动峰,最强振动峰位于595 cm-1处,振动模式表现为2个B原子的剪式振动.
W6B团簇红外光谱在70~230 cm-1范围内集中了许多小峰,最强振动峰位于549 cm-1处,振动模式为B原子的摇摆振动;拉曼光谱主要集中分布在70~300 cm-1频段间,最强峰位于76 cm-1处,振动模式为剪式振动.
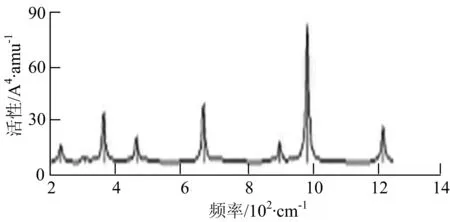
通过对WmBn(m+n≤7)团簇基态结构的光谱分析可知,WmBn团簇的红外和拉曼光谱的振动峰都较多,主要分布在30至1 288 cm-1处,最大频率以及最强峰值对应的振动模式大部分是B原子或W原子的伸缩振动以及摇摆振动.周围环境的改变对频率的变化影响较小,力常数较大致使体系的电偶极矩变化大,所以基团伸缩振动常在高频处出现吸收,W原子之间化学键的力常数较大,容易产生较高的振动频率,所以一般来说频率值较大的几乎都是W原子之间的伸缩振动.WB2,WB3,W2B,W2B2和W2B3团簇红外光谱的吸收峰相对较少,振动模式大部分为伸缩振动.从红外光谱强度图上可以看出,WB5,WB6和W4B2团簇的最强峰分别达到了58.69 km.mol-1,42.20 km.mol-1和40.53 km.mol-1,和其他团簇的最强峰相差较多,说明这两个团簇的振荡模式大大改变了其结构中的电子云分布,偶极距的急剧变化导致了这一现象.
3 结论
采用密度泛函理论中的B3LYP方法,在LANL2DZ基组水平上优化了WmBn(m+n≤7)团簇的几何结构,得到了它们的基态构型,并对其自然键轨道(NBO)以及振动光谱进行了计算研究.对NBO的分析可知,团簇中NBO电荷分布状况与团簇的对称性相关,在对称性较高的团簇中位置相同的原子,其NBO电荷分布状况也相同,W原子比B原子对电荷调节能力强,易和其它原子形成化学键;振荡光谱分析表明,振动频率主要分布在30~1 288 cm-1处,最大频率以及最强峰值对应的振动模式大部分是B原子或W原子的伸缩以及摇摆振动.
[1] Suetin D V, Shein I R, Ivanovskii A L. Structural, electronic properties and stability of tungsten mono-and semi-carbides: a first principles investigation [J].JournalofPhysicsandChemistryofSolids,2009, 70(1): 64-71.
[2] Ding F, Larsson P, Larsson J A, et al. The importance of strong carbon-metal adhesion for catalytic nucleation of single-walled carbon nanotubes[J].NanoLett,2008, 8(2): 463-468.
[3] 张秀荣,高从花,吴礼清,等, WnNim(n+m≤7;m=1,2) 团簇电子结构与光谱性质的理论研究[J].物理学报,2010,59(8):5429-5438.
Zhang Xiurong, Gao Conghua, Wu Liqing,et al.The theory study of electronic structures and spectram properties of WnNim(n+m≤7;m=1,2) clusters[J].ActaPhysicaSinica,2010,59(8):5429-5438.(in Chinese)
[4] Ludwig A, Cao J, Dam B, et al. Opto-mechanical characterization of hydrogen storage properties of Mg-Ni thin film composition spreads[J].AppliedSurfaceScience,2007, 254(3): 682-686.
[5] 仇毅翔,李佳,王曙光.配体稳定的二元过渡金属团簇[PdAu8(PR3)8]2+(R=Me, OMe, H, F, Cl, CN)的量子化学理论研究[J].化学学报, 2010,68(7):611-616.
Qiu Yixiang,Li Jia,Wang Shuguang. Theoretical investigations on ligand-stabilized binary transition-metal cluster [PdAu8(PR3)8]2+(R=Me, OMe, H, F, Cl, CN)[J].ActaChimicaSinica,2010,68(7):611-616.(in Chinese)
[6] Zhang Lin, Zhang Caibei, Qi Yang. Local structure changes of Cu55cluster during heating[J].ChinesePhysics,2007, 16(1): 77-82.
[7] 张秀荣,张伟,高从花,等.(OsB)n(n=1-6)团簇结构与稳定性的第一性原理研究[J]. 计算机与应用化学,2010,27(2):221-224.
Zhang Xiurong,Zhang Wei,Gao Conghua,et al.First-principles study of structures and stable properties of (OsB)n(n=1-6) clusters[J].ComputersandAppliedChemistry,2010,27(2):221-224.(in Chinese)
[8] 华英杰,王崇太,孟长功.NiTi合金表面镍离子释放的电子结构理论计算[J]. 计算机与应用化学,2005,22(12):1115-1118.
Hua Yingjie,Wang Chongtai,Meng Changgong.The theoretical calculation of electronic structure for the release of nickel ion on NiTi alloy surface[J].ComputersandAppliedChemistry,2005,22(12):1115-1118.(in Chinese)
[9] 朱佳,金华,李奕,等.TiO2(110)表面负载W3O10团簇构型和电子结构的理论研究[J].化学学报,2011,69(8):905-911.
Zhu Jia,Jin Hua,Li Yi,et al.Theoretical study on the geometry and electronic structure of W3O10clusters supported on the TiO2(110) surface[J].ActaChimicaSinica,2011,69(8):905-911.(in Chinese)
[10] Yamaguchi W, Murakami J. Geometries of small tungsten clusters[J].ChemicalPhysics,2005, 316(1/3): 45-52.
[11] 姚明珍,顾牡,梁玲,等. PbWO4晶体空位型缺陷电子结构的研究[J].物理学报,2002,51(1):125-128.
Yao Mingzhen,Gu Mu,Liang Ling, et al.Electronic structures of defects associated with intrinsic vacancies in PbWO4crystals[J].ActaPhysicaSinica,2002,51(1):125-128.(in Chinese)
[12] Suetin D V, Shein I R, Ivanovskii A L. Structural, elastic and electronic properties and formation energies for hexagonal (W0.5Al0.5)C in comparison with binary carbides WC and Al4C3from first-principles calculations[J].PhysicaB, 2008, 403:2654-2661.
[13] Wei A, Heath T C. Electronic structure calculations of gas adsorption on boron-doped carbon nanotubes sensitized with tungsten [J].ChemicalPhysicsLetters,2009, 482(4/6): 274-280.
[14] Shane M S, Adam W S, Michael D M. Optical spectroscopy of tungsten carbide WC[J].JournalofChemicalPhysics,2002,116: 993-1002.
[15] 邵泽旭,张启仁,刘延禹,等.CaWO4晶体中F型色心电子结构的研究[J].物理学报,2007,56(7):4089-4093.
Shao Zexu,Zhang Qiren,Liu Yanyu,et al.Electronic structures of CaWO4crystal with F type color center[J].ActaPhysicaSinica,2007,56(7):4089-4093.(in Chinese)
[16] Mannesson K, Elfwing M. Analysis of WC grain growth during sintering using electron backscatter diffraction and image analysis[J].InternationalJournalofRefractoryMetals&HardMaterials,2008,26:449-455.
[17] Tohru Yamasaki. High-strength nanocrystalline Ni-W alloys produced by electrodeposition[J].MaterPhysMech,2000,1:127-132.
[18] 马文瑾,王艳宾,张静,等.BmN (m=2-9)团簇的结构特征与稳定性[J]. 物理化学学报,2007, 23(2): 169-172.
Ma Wenjin,Wang Yanbin,Zhang Jing,et al.Structure characteristics and stability of BmN(m=2-9) clusters[J].ActaPhys-ChimSin,2007, 23(2): 169-172. (in Chinese)
[19] 李莉莎,刘甫,孙久雨,等,AlBn+(n=2~10)团簇结构和红外振动光谱研究[J].光子学报,2011,40(2):321-326.
Li Lisha,Liu Fu, Sun Jiuyu,et al.Theoretical study of structure and infrared vibration spectra about AlBn+(n=2~10) clusters[J].ActaPhotonicaSinica,2011,40(2):321-326. (in Chinese)
[20] 刘火雁,雷雪玲,陈杭,等,FeBN(N≤15)团簇结构、电子性质和磁性的密度泛函理论研究[J].原子与分子物理学报,2011,28(2):258-266.
Liu Huoyan,Lei Xueling,Chen Hang,et al.Structures,electronic properties and magnetisms of FeBN(N≤15) clusters:density functional theory investigations[J].Journalofatomicandmolecularphysics,2011,28(2):258-266. (in Chinese)
[21] 杨致,闰玉丽,赵文杰,等,FeBN(N≤6)团簇的结构与磁性[J].物理学报,2007,56(5):2590-2595.
Yang Zhi,Yan Yuli,Zhao Wenjie,et al.Structures and magnetism of FeBN(N≤6) clusters[J].ActaPhysicaSinica,2007,56(5):2590-2595. (in Chinese)
[22] 张秀荣,刘小芳,康张李.W6Sin0,±(n=1,2)团簇结构与电子性质的密度泛函理论研究[J].江苏科技大学学报:自然科学版,2010,24(6):619-624.
Zhang Xiurong,Liu Xiaofang,Kang Zhangli.Density functional theory study on the structure and electronic properties of W6Sin0,±(n=1,2) clusters[J].JournalofJiangsuUniversityofScienceandTechnology:NaturalScienceEdition,2010,24(6):619-624. (in Chinese)
[23] 张秀荣,李扬,杨星. WnNim(n+m=8)团簇结构与电子性质的理论研究[J]. 物理学报, 2011,10:236-245.
Zhang Xiurong,Li Yang,Yang Xing.Theoretical study on structural and electronic properties of WnNim(n+m=8) clusters[J].ActaPhysicaSinica,2011,10:236-245. (in Chinese)
猜你喜欢
数学物理学报(2022年3期)2022-05-25
数学物理学报(2022年1期)2022-03-16
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
数学物理学报(2021年5期)2021-11-19
数学物理学报(2021年3期)2021-07-19
中国粮油学报(2018年12期)2018-03-19
国外科技新书评介(2014年8期)2014-12-05
