
Synthesis and Crystal Structure of (Z)-N-(2-(diethylamino)ethyl)-7-(5-fluoro-2-oxoindolin-3-ylidene)-2-methyl-4,5,6,7-tetrahydro-1H-indole-3-carboxamide Methanol Solvate①
2013-10-11 03:00:14JINQiuYOUQiDongTANGFengDrugDesignndPtentMedicineOptimiztionKeyLortoryofJingsuProvinceChinPhrmceuticlUniversityNnjing210009ChinTrgetingAntitumorDrugKeyLortoryofJingsuProvinceSimcerePhrmceuticlGroupofJingsuProvinceNnjin
结构化学 2013年7期
JIN Qiu YOU Qi-Dong TANG Feng② (Drug Design nd Ptent Medicine Optimiztion Key Lortory of Jingsu Province, Chin Phrmceuticl University, Nnjing 210009, Chin) (Trgeting Antitumor Drug Key Lortory of Jingsu Province,Simcere Phrmceuticl Group of Jingsu Province, Nnjing 210042, Chin)
1 INTRODUCTION
Oxindole is an important type of heterocyclic compound, whose structure can be found in many drug molecules, such as sunitinib. After the appearance of sunitinib on the market, a series of oxindole-based antineoplastic drugs has been developed by some pharmaceutical companies. (Z)-N-(2-(diethylamino)ethyl)-7-(5-fluoro-2-oxoindolin-3-ylidene)-2-methyl-4,5,6,7-tetrahydro-1H-indole-3-carbox amide (compound 3) is a new oxindole-based compound, which is proposed according to the structure-property relationship of sunitinib. It exhibits activity inhibition effect against many tyrosine kinases (including VEGFR-2, PDGFR-β and c-Kit),and can treat many types of tumors potentially[2].
Synthesis route of compound 3 is shown in Fig. 1.2-methyl-7-oxoindolin-4,5,6,7-tetrahydro-1H-indole-3-carboxylic acid (compound 1) reacts with N,N-diethyl-1,2-ethanediamine to produce N-2-(diethylamino)ethyl)-2-methyl-7-oxoindolin-4,5,6,7-tetrahy dro-1H-indole-3-carboxamide (compound 2) via condensation reaction, and then 2 reacts with 5-fluoro-1H-indole-2-ketone to generate compound 3 under the Lewis acid catalyst[2]. Compound 3 was characterized by NMR, MS and elemental analysis.Furthermore, the crystal structure of its methanol solvate was determined by X-ray diffraction method.

Fig. 1. Synthesis route of (Z)-N-(2-(diethylamino)ethyl)-7-(5-fluoro-2-oxoindolin-3-ylidene)-2-methyl-4,5,6,7-tetrahydro-1H-indole-3-carboxamide (1-3)
2 EXPERIMENTAL
2.1 Reagents and physical measurements
All chemicals were purchased from commercial resources and used without any further purification.1H- and13C-NMR spectra were recorded on a BRUKER AV-500 NMR spectrometer using CDCl3and DMSO-d6as solvents and TMS internal label.MS measurement was carried out on an Agilent G1956B single quadrupole mass spectrometer(ionization model: ESI, solvent (80): methanol:water(20)). Elemental analysis was performed on an ElementarVario EL III elemental analysis instrument.
2.2 Synthesis and characterization
2.2.1 Synthesis of compound 2
Compound 1 (0.20 g, 1.0 mmol), 0.16 g HOBt(1.2 mmol), 0.24 g (1.2 mmol) EDCI and 0.20 g (2.0 mmol) triethylamine were dissolved in 10 mL DMF.After stirring at room temperature for 20 min, 0.24 g(2.1 mmol) N,N-diethyl-1,2-ethanediamine was added and kept reacting at room temperature for 24 h. Afterwards, the resultant solution was moved into ice water and the product was extracted with CH2Cl2.The organic layer was washed with saturated NaCl solution and dried with anhydrous sodium sulfate,and the white solid (compound 2) treated by condensation was purified by silica column chromatography method (eluant: CH2Cl2:methanol = 30:1,v:v). Yield: 0.23 g, 79%.
1H NMR (500 MHz, CDCl3) δ: 9.67(s, 1H,-NH-1), 6.41(s, 1H, -CONH-), 3.46(bs, 2H,-CONHCH2-), 2.93(t, J = 9.97 HZ, 2H, -CH2-4),2.64~2.59(m, 6H, -NHCH2CH3)2), 2.58(s,3H, -CH3-2), 2.49(t, J = 8.92 HZ, 2H, -CH2-6),2.16(m, 2H, -CH2-5), 1.04(s, 6H, -(CH2)2);ESI-MS: 292.2 [M+H]+, 314.2[M+Na]+; 290.3[M-H]-.
2.2.2 Synthesis of compound 3
0.12 g (0.79 mmol) 5-fluoro-2-hydroxyl-indole and 0.20 g (0.69 mmol) compound 2 were dissolved in 10 mL pyridine, and then 0.2 mL TiCl4was added and kept reacting at 100~110 ℃ under stirring for 10 h. The resultant solution was poured to ice water and the product was extracted with CH2Cl2. The organic layer was washed with saturated NaCl solution and dried with anhydrous sodium sulfate,and the yellow solid (compound 3) treated by condensation was purified by silica column chromatography method (eluant: CH2Cl2:methanol = 30:1,v:v). Yield: 0.09 g, 31%.
1H NMR(500MHz, DMSO-d6) δ: 14.55(s, 1H,-NH-), 10.87(s, 1H, -NH-), 7.39(dd, J = 2.30, J =10.95 HZ, 1H, PhH), 7.19(t, J = 5.45 HZ, 1H,-CONH-), 6.93(td, J = 2.35, J = 8.80 HZ, 1H, PhH),6.86(dd, J = 5.05, J = 8.50 HZ, 1H, PhH), 3.30(q, J= 6.45 HZ, 2H, -CONHCH2-), 3.01(t, J = 6.00 HZ, 2H, -CH2-), 2.83(t, J = 6.00 HZ, 2H, -CH2-),2.50~2.56(m, 6H, -NHCH2N(CH3)2), 2.46(s,3H, -CH3), 1.94(t, J = 5.85 HZ, 2H, -CH2-), 0.97(t, J= 7.00 HZ, 6H, -(CH213C NMR(125MHz,DMSO-d6) δ: 169.42(s), 164.19(s), 158.46(d, J =231.25), 145.49(s), 136.37(s), 134.73(s), 132.74(s),126.19(d, J = 8.75), 125.52(s), 117.63(s), 111.82(d, J= 2.5), 111.45(d, J = 23.75), 110.63(d, J = 26.25),109.43(d, J = 8.75), 51.60(s), 46.46(s), 36.89(s),30.56(s), 23.52(s), 22.45(s), 13.32(s), 11.85(s);ESI-MS: m/z = 425.1 [M+H]+. Elemental analysis Calcd. for C24H29FN4O2: C, 67.90; H, 6.89; F, 4.48;N, 13.20. Found (%): C, 67.86; H, 7.16; F, 4.58; N,13.01.
2.3 X-ray structure determination
Single crystals of the title compound were obtained by the following steps: the recrystallized title compound was dissolved in methanol in a 100 mL conical flask, and the solution was evaporated at room temperature to get single crystals.
A crystal with dimensions of 0.10mm × 0.20mm ×0.30mm was selected[3-7]and measured on an Enraf-Nonius CAD4 EXPRESS diffractometer equipped with a graphite-monochromatized MoKα radiation (λ = 0.71073 Å) for data collection at 293±1 K with an ω-2θ scan method. A total of 4262 independent reflections were collected in the range of 1.4<θ<25.4° and 2701 were observed with I >2σ(I). The structure was dissolved by direct methods.Non-hydrogen atoms were determined with successive difference Fourier syntheses. The hydrogen atoms were located at the calculated positions. The anisotropic thermal parameters for non-hydrogen atoms were refined by full-matrix least-squares of F2.All calculations were performed on a computer with SHELX 97 program package. The final R = 0.0604,wR = 0.1644 (w = 1/[σ2(Fo2) + (0.0721P)2+0.9935P], where P = (Fo2+ 2Fc2)/3), S = 1.02,(Δρ)max= 0.34 and (Δρ)min= –0.23 e/Å3. Crystal parameters: monocline system, space group P21/c, a= 14.560(3), b = 7.320(2), c = 22.233(4) Å, β. =101.78(3)°, Z = 4, V = 2319.7(8) Å3, Dc= 1.307 g/cm3, F(000) = 976 and Mr= 456.55.
3 RESULTS AND DISCUSSION
Selected bond lengths and bond angles together with torsion angles are given in Table 1. The molecular structure showing atomic labeling scheme can be seen in Fig. 2, and its packing diagram is revealed in Fig. 3. According to the structural analysis, the bond lengths of C(4)–C(5), C(5)–C(6),C(7)–C(9), C(13)–C(14) and C(15)–C(16) are obviously longer than that of the normal C=C double bond (1.34 Å), and the corresponding C(6)–N(1),C(8)–N(1), C(14)–N(2), C(16)–N(2), C(18)–N(3),C(5)–C(7), C(7)–C(8), C(9)–C(14), C(13)–C(15)and C(15)–C(18) bond lengths are shorter than those of C(sp2)–N (1.426 Å) and C–C (1.53 Å). So, a delocalization to some extent has occurred, suggesting that a larger conjugated system defined by indole ring, pyrrole ring and carbonyl has been produced.

Table 1. Selected Bond Lengths, Bond Angles (Å) and Torsion Angles (°)
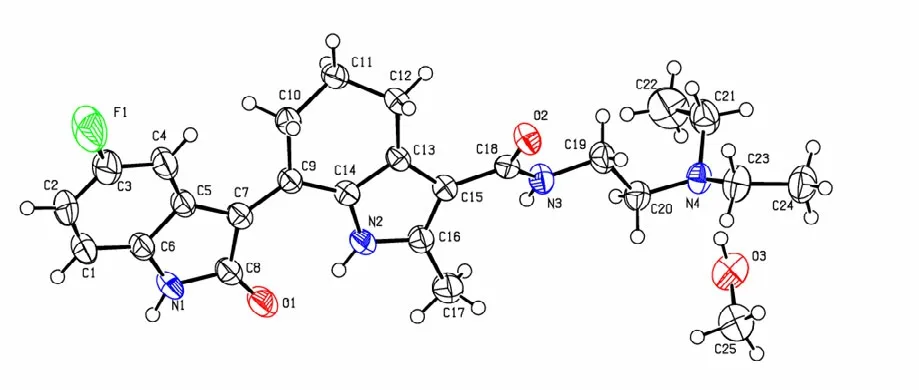
Fig. 2. Molecular structure of (I), showing the atomic labeling scheme

Fig. 3. Packing view of the title compound, showing the hydrogen bonds (dashed lines) and π-π stacking interactions
In the indolone ring, N(1)/C(1)/C(2)/C(3)/C(4)/C(5)/C(6)/C(7)/C(8) is generally coplanar. Among with C(3) and C(5) exhibit the largest deviations of–0.044(2) and 0.050(2) Å. Pyrrole ring(N(2)/C(14)/C(13)/C(15)/C(16)) shows better coplanarity (N(2) and C(16)), showing the largest deviations of –0.006(2) and 0.005(2) Å, respectively). Indolone and pyrrole rings are generally coplanar with the dihedral angle of 8.13(6)°.
The puckering amplitude of cyclohexene ring(C(9)/C(10)/C(11)/C(12)/C(13)/C(14)) is 0.471(2) Å,illustrating its envelope-type configuration (Phi is 123.0(5)°).
Judging from the packing diagram of the title compound (Fig. 3), intra- and intermolecular hydrogen bonds of C–H··O and N–H··O contribute to the structural stabilization (Table 2). Besides, weak π-π packing interactions between Cg2 (pyrrole ring N(2)/C(14)/C(13)/C(15)/C(16)) and Cg3 (benzene ring C(1)~C(6)) can be observed (Cg2–Cg3 distance:3.48~3.80 Å, centroid distance: 3.96~4.00 Å,symmetry codes: 1–x,–1/2+y,1/2–z, 1–x, 1/2+y,1/2–z). A 1-D chain along the c-axis was given upon the intermolecular hydrogen bonding interactions.Furthermore, the 1-D chain extends into a 2-D layer via π-π packing interaction and intermolecular hydrogen bonds, and the crystal structure is thus stabilized.

Table 2. Hydrogen Bonding Interactions (Å, °)
(1) Madhusudan, S.; Ganesan, T. S. Tyrosine kinase inhibitors in cancer therapy. ClinBiochem. 2004, 37, 618–635.
(2) Tang, F.; Shen, H.; Jin, Q. 3-Pyrrolo-cyclohexylene-2-dihydro-indolinone derivatives and uses thereof. WO2008067756A1 2008.
(3) Bukovskii, M. I.; Solodushenkov, S. N.; Mosiichuk, A. I. (Phosphoranylideneamino)-s-triazinesⅢ . Oxidative imination of tervalent phosphorus compounds with azido-s-triazines. Translated from ZhurnalObshcheiKhimii 1970, 40, 782–784.
(4) Wu, Z. Y.; Xu, W.; Xia, J. K.; Liu, Y. C.; Wu, Q. X.; Xu, W. J. Flame retardant polyamide 6 by in situ polymerization of -caprolactam in the presence of melamine derivatives. Chin. Chem. Lett. 2008, 19, 241–244.
(5) Alexej, B. B.; Albert, B. D. Insect chemosterilants. Ⅴ . Derivatives of melamine.Insect Chemosterilants 1967, 10, 457–461.
(6) Tatyana, S.; Christian, L.; Uwe, B.et al. Melem- and melamine-derived iminophosphoranes. New Journal of Chemistry 2010, 34, 1893–1908.
(7) X-ray Single-crystal Analysis System. Version 2.2, Bruker AXS Inc., 6300 Enterprise Lane, Madison, WI 53719–1173, USA.

- 结构化学的其它文章
- Synthesis and Crystal Structure of 5,5΄-Bis(4-hydroxylphenyl)diazo-dipyrromethane①
- A Novel Cu-based Metallosalan Complex:Synthesis, Structure and Chiral Sensor Study①
- Syntheses, Crystal Structures and Properties of Two Metal-organic Complexes Based on 5-(4-Pyridyl)-methoxyl Isophthalic Acid①
- DFT Study on the Electronic and Structural Properties of MoS6-/0 Clusters①
- Synthesis, Crystal Structure, and Biological Activity of 4-Chlorobenzaldehyde(2-trifluoromethylTrifluoromethyl-5,6,7,8-tetrahydrobenzo[4,5]- thieno[2,3-d]pyrimidin-4-yl)hydrazone Monohydrate①
- Interesting Weak Interactions in Three Silver Coordination Polymers Based on Tetrafluorobenzenedicarboxylate and Benzonitrile Ligands①