
Synthesis of a Weak Basic uPA Inhibitor and Crystal Structure of a Complex with uPA①
2013-10-11 03:00:10YUHiYngGAODnZHANGXuJIANGLongGungHONGZeBinFANGXinYUANCiWANGJunDongHUANGMingDongCollegeofChemistryndChemiclEngineeringFuzhouUniversityFuzhou350108ChinStteKeyLortoryofStructurlChemistryFujinInstituteofReserchonthe
结构化学 2013年7期
YU Hi-Yng GAO Dn ZHANG Xu JIANG Long-Gung HONG Ze-Bin FANG Xin YUAN Ci WANG Jun-Dong HUANG Ming-Dong, ② (College of Chemistry nd Chemicl Engineering,Fuzhou University, Fuzhou 350108, Chin) (Stte Key Lortory of Structurl Chemistry, Fujin Institute of Reserch on the Structure of Mtter, Chinese Acdemy of Sciences, Fuzhou 350002, Chin)
1 INTRODUCTION
uPA is a trypsin-like serine protease which plays an important role in various biological processes[1].uPA activates plasminogen to plasmin which in turn initiates the activation of many matrix metalloproteases. Besides as a key initiator in extracellular proteolysis, uPA is also a key mediator in a range of biological processes including angiogenesis, wound healing, tissue remodeling, tumor growth and tumor metastasis[2]. Accordingly, selective inhibitor for uPA will have therapeutic values in cancer and wound healing, and has been a focus of intensive research for decades[1,3-4]. Several uPA inhibitors with high potency and selectivity were invented in the last decade, and these inhibitors were shown to have potential anticancer effects[5-6]. A common feature of such inhibitors is the presence of a guanidine or an amidine moiety (named here as P1 group) in these inhibitors which insert into the S1 specific pocket of uPA and interact with the conserved residue Asp189 (chymotrypsinogen numbering). However, the highly basic amidine or guanidine group inevitably conduces poor bioavailability of the uPA inhibitor[7]. Thus, the design of uPA inhibitors with less basic P1 group will be highly desirable and may have improved bioavailability.
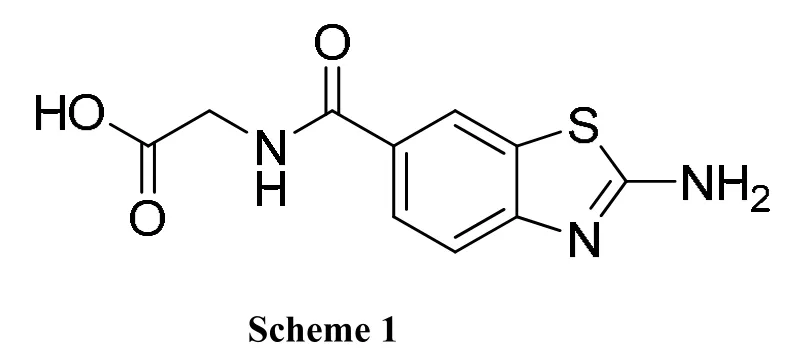
In this study, 2-aminobenzothiazole (ABT), a weakly basic group with pKa of 4.51[8], was used as P1 group for uPA inhibition. We synthesized a new small molecule 2-(2-aminobenzothiazole-6-carboxamido) acetic acid (ABTCA, Scheme 1). This small molecule inhibited uPA with an efficacy about 20-fold higher than ABT. In addition, we determined the crystal structure of uPA in complex with ABTCA. The crystal structure showed that the ABT moiety of inhibitor indeed binds to the S1 pocket of uPA, and acetic acid moiety occupies the oxyanion hole.
2 EXPERIMENTAL
2.1 Chemistry
The starting compound, 2-aminobenzothiazole-6-carboxylate, was commercially purchased without further purification. Solvents used in this study were used without further purification except tetrahydrofuran (THF) was re-distilled. Mass spectra were measured on the Thermo Finnigan DECAX-30000 LCQ Deca XP spectrometer (ESI-MS).1H NMR spectra were determined with a Bruker Biospin AVANCE III NMR spectrometer operating at 400 MHz. The synthesis procedure and conditions are shown in Scheme 2.
2.1.1 Synthesis of ethyl 2-(tert-butoxycarbonylamino)benzothiazole-6-carboxylate (1)
The mixture of 2-aminobenzothiazole-6-carboxylate (444 mg, 2 mmol), 4-dimethylamino-pyridine(48 mg, 0.4 mmol) and diisopropylethylamine (520 μL, 3 mmol) in 15 mL of THF was stirred and added dropwise into a solution of di-tert-butyl dicarbonate(523 mg, 2.4 mmol) in 15 mL of THF under room temperature. The reaction was continued until thin layer chromatography (TLC, hexane/ethyl acetate:V/V = 2:1) showed the disappearance of 2-aminobenzothiazole-6-carboxylate. Then, the solvent was removed with vacuum. The residual was dissolved in dichloromethane (DCM). The solution was washed with 0.5 N HCl and 5% Na2CO3solution, brined and dried with anhydrous Na2SO4. Then the solvent was removed under reduced pressure to give 601 mg white solid (compound 1) in 93% yield. ESI-MS:m/z, calcd (M) 322.1, found 321.5 (M-, 100%).1H NMR (400 MHz, δ, DMSO-d6): 12.00 (s, 1H); 8.59(d, 1H, J = 1.2 Hz); 7.98 (dd, 1H, J1= 1.6 Hz, J2=8.4 Hz); 7.75 (d, 1H, J = 8.8 Hz); 4.34 (m, 2H);1.53 (s, 9H); 1.34 (t, 3H).
2.1.2 2-(Tert-butoxycarbonylamino)benzothiazole-6-carboxylic acid (2)
A mixture of compound 1 (644 mg, 2 mmol) and NaOH (600 mg, 15 mmol) in 15 mL of methanol/H2O (V/V = 1:1) was stirred and heated to reflux until TLC showed the disappearance of compound 1 (hexane/ethyl acetate: V/V = 2:1). Then the solution was added into 50 mL of water and extracted with ethyl acetate. When the water layer was soured with 3 M HCl to pH ≈ 4, a precipitate formed. The precipitate was filtered, washed with water until the pH of the filtrate reached about 7, and dried to give white solid 570 mg, yield: 97%. ESI-MS: m/z, Calcd. (M) 294.1, found 293.5 (M-, 100%).1H NMR (400 MHz, δ, DMSO-d6): 12.00 (s, 1H),8.56 (d, 1H, J = 1.2 Hz), 7.96 (dd, 1H, J1= 1.6 Hz, J2= 8.4 Hz), 7.72 (d, 1H, J = 8.8 Hz), 1.52 (s, 9H).
2.1.3 Methyl 2-(2-(tert-butoxycarbonylamino)benzothiazole-6-carboxamido)acetate (3)
The mixture of compound 2 (588 mg, 2 mmol),2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU, 758 mg, 2 mmol) and N,N-diisopropylethylamine (DIPEA, 525 μL, 3 mmol) in 15 mL of N,N-dimethylformamide (DMF)was stirred for 1 h under room temperature. Then to the mixture was added glycine methyl ester hydrochloride (301 mg, 2.4 mmol) and 525 μL of DIPEA(3 mmol), followed by stirring overnight. The solution was added into water (150 mL) and extracted with ethyl acetate (100 mL × 3). Combined organic layer was washed by 0.5 M HCl, 5%Na2CO3solution, and saturation brine, and dried over anhydrous Na2SO4. The solution was concentrated under reduced pressure to about 10 mL,leading to precipitation. The precipitate was filtered,washed with ethyl acetate, and dried to give white solid 548 mg, yield: 75%. ESI-MS: m/z, Calcd. (M)365.1, found 364.4 (M-, 100%);1H NMR (400 MHz,δ, DMSO-d6): 8.98–8.95 (t, 1H, J = 5.8 Hz),8.45–8.44 (d, 1H, J1= 1.6 Hz), 7.92–7.89 (dd, 1H, J1= 2 Hz, J2= 8.4 Hz). 7.74–7.72 (d, 1H, J = 8.8 Hz),4.04–4.03 (d, 2H, J = 3.6 Hz), 3.66 (s, 3H), 1.52 (s, 9H).
2.1.4 2-(2-(Tert-butoxycarbonylamino) benzothiazole-6-carboxamido)acetic acid (4)
A mixture of compound 3 (730 mg, 2 mmol) and LiOH·OH (168 mg, 4 mmol) in 15 mL of THF/H2O(V/V = 1:1) was stirred under room temperature until TLC showed the disappearance of compound 1(hexane/ethyl acetate: V/V = 1:2). Then the solution was added to 50 mL of 0.1 M NaOH solution and extracted with ethyl acetate. The water layer was soured with 3 M HCl to pH ≈ 4, giving a precipitate.The precipitate was filtered, washed with water, and dried to give white solid 568 mg, yield: 81%.ESI-MS: m/z, Calcd. (M) 351.1, found 350.4 (M-,100%);1H NMR (400 MHz, δ, DMSO-d6):8.8.89–8.85 (t, 1H, J = 5.8 Hz), 8.47–8.46 (d, 1H, J1= 1.6 Hz), 7.94–7.91 (dd, 1H, J1= 2 Hz, J2= 8.4 Hz).7.75–7.73 (d, 1H, J = 8.8 Hz), 3.97–3.96 (d, 2H, J =4 Hz), 1.53 (s, 9H).
2.1.5 2-(2-Aminobenzothiazole-6-carboxamido)acetic acid trifluoroacetate (ABTCA·TFA)
The mixture of compound 4 (200 mg, 0.57 mmol),0.3 mL of DCM, and 1.7 mL of trifluoroacetic acid was stirred for 4 h under room temperature. The solution was added into 30 mL of ethyl ether to give a precipitate. The precipitate was filtered, washed with ether, and dried to give white solid 176 mg,yield: 85%. ESI-MS: m/z, Calcd. (M) 251.0, found 250.7 (M-, 100%).1H NMR (400 MHz, δ,DMSO-d6): 8.80–8.77 (t, 1H, J = 5.6 Hz), 8.34 (s,2H), 8.29 (d, 1H, J1= 1.2 Hz), 7.87–7.84 (dd, 1H, J1= 1.6 Hz, J2= 8.4 Hz). 7.47–7.45 (d, 1H, J = 8.4 Hz),3.99–3.97 (d, 2H, J = 6 Hz), 1.53 (s, 9H).
2.2 Expression and purification of recombinant uPA protease domain (16-244)
The uPA protease domain (Ile16-Glu244 in the chymotrypsin numbering) with C122A and N145Q was cloned into the pPICZa expression vector according to Zhao et al[9]. The recombinant uPA was secreted from transfected pichia pastoris X-33 cells after induction by methanol. The recombinant uPA was captured from the expression medium by a cation exchange column SPFF and eluted with a NaCl gradient (0~0.5 M, 300 mL) in 20 mM phosphate buffer, pH = 6.5. The eluent was concentrated to 4 mL by Millipore ultra-filtration tube and then applied to a gel filtration chromatography(Superdex75 HR 10/30 column) equilibrated with 20 mM phosphate buffer, pH 6.5, containing 150 mM NaCl. The protein was eluted as a single peak with a retention time of approximately 13.6 mL and was confirmed to be uPA by SDS-PAGE analysis (molecular weight of 28000) under reducing condition.The protein was dialyzed in 20 mM potassium phosphate at pH 6.5 overnight and concentrated to 10 mg/mL, using stirred ultra filtration cells (Millipore and micon Bioseparations, Model-5124) for crystallization.
2.3 Enzyme inhibition assays
The inhibitors at various concentrations were incubated with uPA (10 nM) in phosphate buffer saline (20 mM phosphate, 150 mM NaCl, pH 7.4)containing 0.1% BSA at room temperature for 15 min. Chromogenic uPA substrate H-D-pyroglutamyl-Gly-L-Arg-p-nitroanilide (S-2444, Chromogenix-instrumentation Laboratories, Milano, and TM)at 0.16 mM was added into a total volume of 100 μL,and absorbance at 405 nm was then recorded for 15 min using a BioTek Synergy 4 automatic microplate reader. Background values were subtracted from the final absorbance values. Percentage inhibition was calculated and plotted against compound concentrations to generate IC50 values. The inhibitory constants (Ki) were calculated from known Km of the substrate, 90 μM, using the following equation[10]:Ki = IC50/[1 + ([S]/Km)].
2.4 Crystallization, X-ray data collection, and structure determination of uPA-inhibitor complexes
The crystals of uPA were obtained by vapor diffusion method as described previously[9]. Inhibitors were then soaked into the crystals in mother liquor consisting of 0.05 M sodium citrate (pH 4.6), 1.95 M(NH4)2SO4, 0.05% NaN3and 5% PEG400. X-ray data set of the ABTCA-uPA complex was collected to 2.24 Å in our laboratory using a Bruker Smart 6000 charge-coupled device detector equipped with a Nonius FR591 X-ray generator and Cu rotating anode. Prior to mounting on the X-ray machine, the crystals were dipped briefly in the mother liquor containing 25% glycerol (v/v) as cryoprotectant. The structures of uPA-ABTCA complexes were solved by molecular replacement[11]and refined by CNS package[12]. Inhibitor was then modeled into the largest positive peaks of Fo–Fc difference maps. The structure was then refined by maximum-likelihood minimization, followed by individual B-factor refinement[12]. Water molecules were added, using the molecular graphics program COOT[13], and the structure was refined until convergence (Table 1).The final structures were analyzed by software Pymol[14]. X-ray diffraction data and the final refinement data are shown in Table 1.
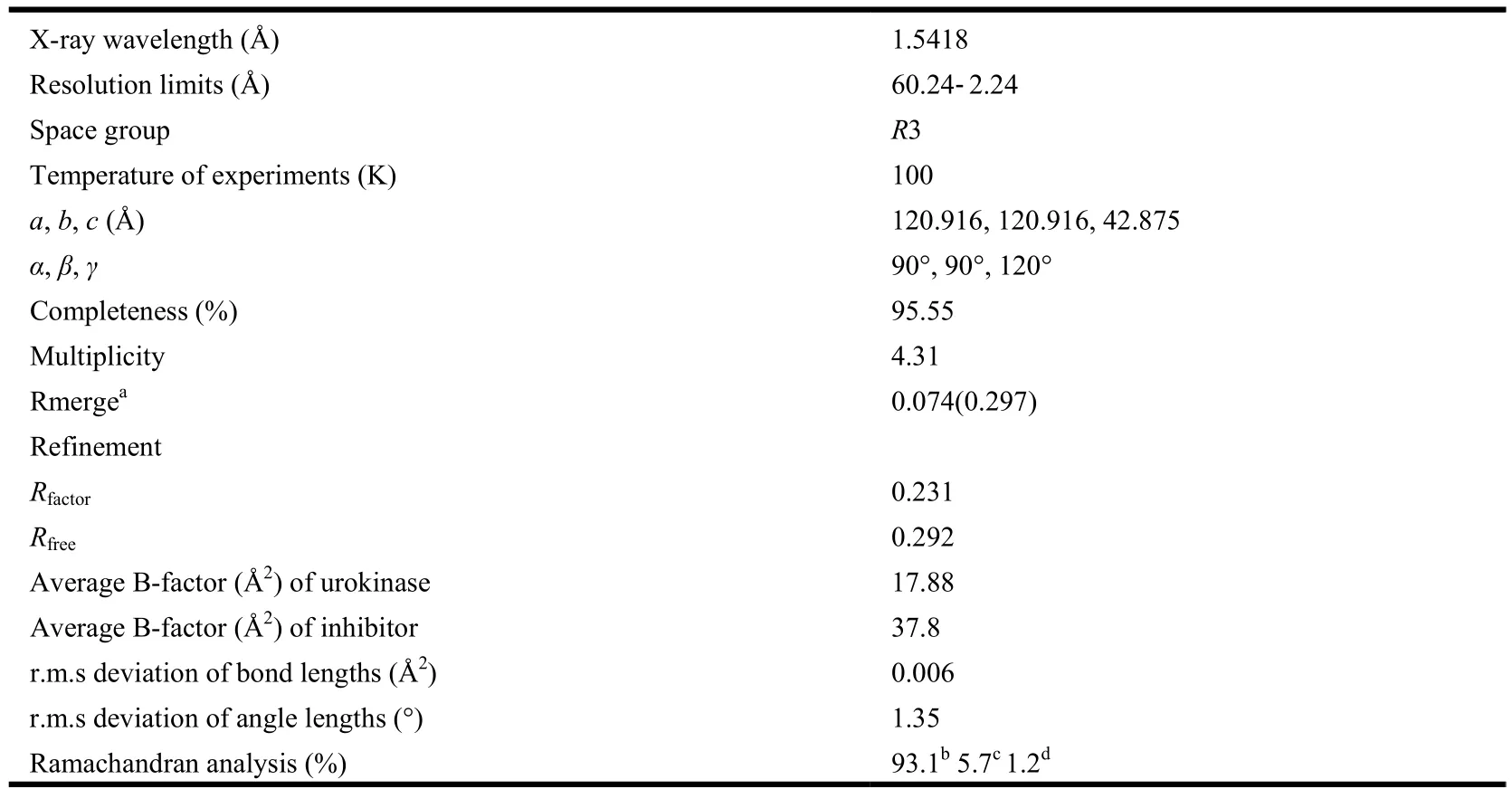
Table 1. X-ray Data Collection and Model Refinement Statistics
3 RESULTS AND DISCUSSION
3.1 Synthesis
We use ethyl 2-aminobenzothiazole carboxylate as template to design uPA inhibitor because this compound is commercially available and has welldefined and feasible synthetic routes to produce[15-17].The 2-aminobenzothiazole can be used to target the S1 specific pocket of uPA protease, and has weak basicity and thus may have higher bioavailability. In this report, we synthesize ABTCA via a five-step synthetic method with good total yield of about 50%.
All compounds in this synthesis process were characterized by MS and H1NMR.
3.2 Biological activity
The ABTCA compound inhibited the uPA with Ki of 341 μM using a fluorogenic competitive enzy-matic assay. This inhibitory efficacy toward uPA is about 20 folds higher compared to ABT (Ki of 5.03 mM)[18], most likely contributed by side chain on the benzothiazole ring.
3.3 Crystal structure of uPA-ABTCA complex
To study the structural mechanism of uPA inhibition by ABTCA, we determined the crystal structure of uPA-ABTCA complex to a resolution of 2.01 Å. The structure was refined to R and Rfreevalues of 0.219 and 0.293, respectively, and satisfactory stereochemistry with 93.1% residues in the favorable Ramachandran dihedral angles (Table 1). The structure shows that the 2-aminobenzothiazole moiety of ABTCA binds to the S1 specific pocket of uPA, and makes extensive hydrogen bonds to uPA either directly or through a water molecule (Table 2).
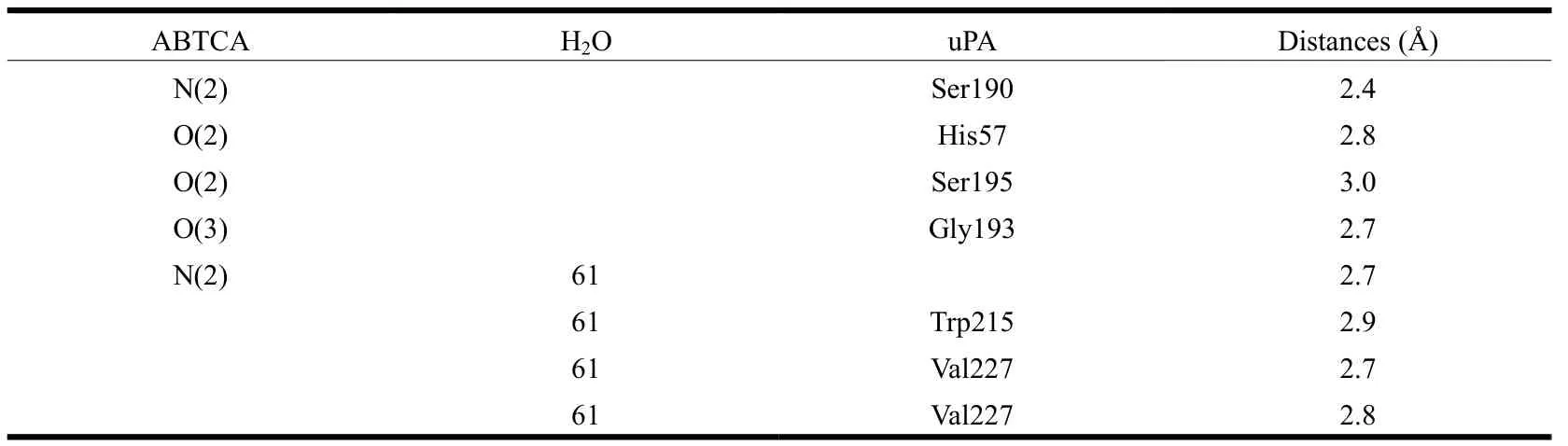
Table 2. Hydrogen Bonds between uPA and ABTCA
Structure of ABTCA-uPA complex reveals that the 2-aminobenzothiazole moiety of ABTCA inserts to the S1 pocket (Fig. 1). However, it is quite interesting to note that no H-bond is formed between the amino group oxygen atoms of ABTCA and Asp189 at the bottom of S1 pocket: the distances between the amine nitrogen atom (N2) of ABTCA and the carboxyl oxygen atoms of Asp189 are 3.8 and 4.4 Å, respectively. This is unusual considering the charge complementarity of the amine group of ABTCA and the uPA Asp189 residue. Instead, a hydrogen bond with 2.4 Å is formed between the amine group and O atom of Ser190 residue. Additionally, the amine group interacts with the main chain oxygen of Trp215, the main chain oxygen and nitrogen of Val227 bridged by a water molecule. The distance between the amine group and the water molecule is 2.7 Å. And the distances from water molecule to the O atom of Trp215, the O and N atoms of Val227 are 2.9, 2.7, and 2.8 Å, respectively.
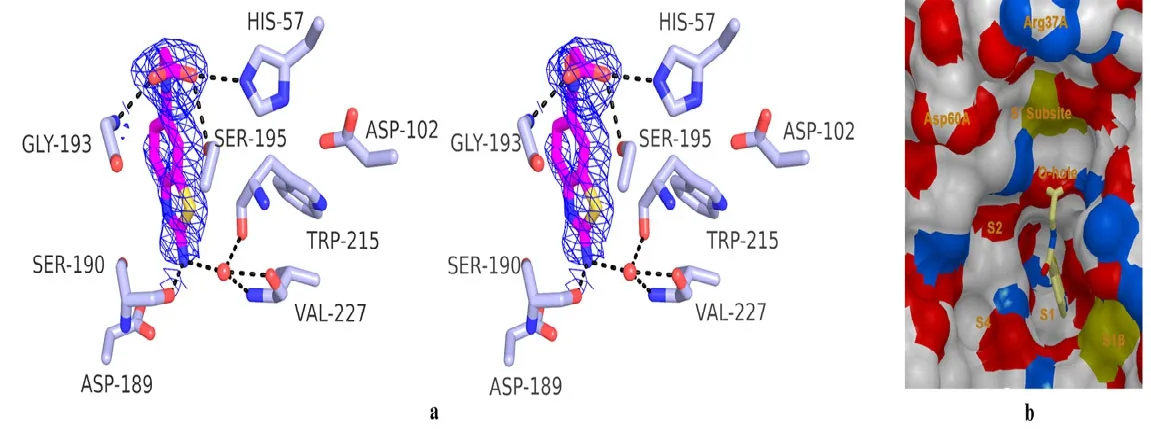
Fig. 1. Crystal structure of the catalytic domain uPA in complex with 2-(2-aminobenzothiazole-6-carboxa mido)acetic acid (ABTCA). a) Stereoview of uPA (gray sticks) in the complex with ABTCA (purple sticks).The 2Fo-Fc density map for the ABTCA ligand is shown as blue. Selected uPA residues discussed in the text(His57, Asp102, Asp189, Ser190, Gly193, Ser195, Val227, Trp215) and an important water molecule (water 61)are shown. b) Surface representation of specific pockets and S′ subsite of uPA with ABTCA shown as the sticks
The structure also shows additional hydrogen bonds between the uPA and carboxyl group moiety of ABTCA (Fig. 1). The carboxyl group occupies the characteristic oxyanion hole (main chain N atom of Gly193 and Ser195). This oxyanion hole is important for the hydrolysis of substrate and is typically occupied by a bound sulfate ion originating from the crystallization buffer in the crystal structures of the complexes between uPA and many small molecular inhibitors[19-21]. The O(2) atom of carboxyl group directly contacts with the side chain of Ser195 and His57 through H-bond, with distances of 2.8 and 3.0 Å, respectively. In addition, the O(3) atom in the carboxyl group interacts with Gly193 via H-bond with a distance of 2.7 Å. Thus, it is these interactions between the carboxyl group and uPA that render the 20-fold higher inhibition of ABTCA compared to ABT.
3.4 Discussion
Low bioavailability is a key challenge faced in the development of protease inhibitors. This is mainly due to the typical use of guanidine or amidine groups in the inhibitors (P1 group) to guide them to the S1 pocket by interacting to the conserved aspartate (Asp189). We propose to use less basic 2-aminobenzothiazole groups as P1 group to target at the S1 pocket. In this report, we synthesized ABTCA and showed it indeed can inhibit uPA. We further determined the crystal structure of uPAABTCA complex to high resolution. To our surprise,the structure showed that the amine group is only bound to Ser190 but not Asp189 (Fig. 1).
As a typical serine protease, uPA structure[22]revealed similarities with other trypsin-like serine proteases[23-27]which include the catalytic triad (Ser195,His57, Asp102) and the deep S1 pocket that has a negatively charged residue (Asp189) at its base.However, the residue at position 190, adjacent to the Asp189, is a serine for uPA, trypsin and plasmin,whereas tPA, thrombin and factor Xa have an alanine at this position. Thus, the direct interaction between ABTCA and Ser190 will likely offer specificity of ABTCA to uPA, which will need further experimental test.
Why does ABTCA not insert into the S1 pocket deeper and interact with the Asp189? The reasons may be that the base of ABT is too weak, or the S1 pocket is not large enough to allow the benzothiazole to fill deeper, or there exists interaction between the periphery groups of ABTCA and uPA non-S1 pocket (exosites). In our previous study, we found that the ABT was able to enter the S1 pocket deeper and interact with the Asp189 directly[18],ruling out the possibility that ABT is too bulky to penetrate into the S1 pocket (Fig. 2). Is weak basicity of ABT the reason preventing ABTCA from burying deeper in the S1 pocket? This seems not a strong reason because even a highly basic amidino group did not interact directly with the Asp189[28].Therefore, we may conclude that the interactions between the carboxyl group of ABTCA and the uPA oxyanion hole must be quite strong to an extent preventing the deeper penetration to the S1 pocket and the inhibitor interaction with the Asp189. Thus,it may be due to this lack of interaction to Asp189 that accounts to the relatively low Ki of ABTCA.
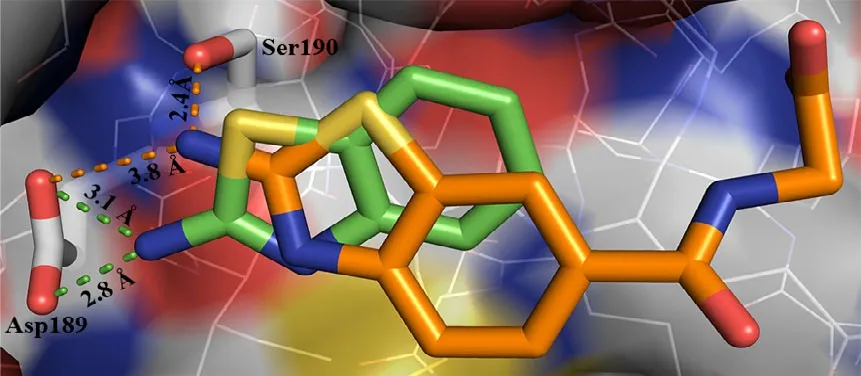
Fig. 2. An overlay of ABTCA (orange) with ABT (green)
The current study sheds light on the further design of uPA inhibitors. If one could design a molecule with both exosite interaction and the interaction to the Asp189, the molecule would have high inhibitory efficacy toward uPA. Another strategy to increase the inhibitory efficacy and at the same time the specificity is to target both the S1 pocket and the so-called 37- and 60-loops, which are located farther away from the S1 pocket. These loops are quite different among various serine proteases (Table 3).We are currently working on these approaches.
4 CONCLUSION
We synthesized a new uPA inhibitor, 2-(2-aminobenzothiazole-6-carboxamido)acetic acid, with Ki of 341 μM, and delineated the binding mechanism by determining the crystal structure of uPA-inhibitor complex. The structure information is important for further improvement of uPA inhibitor with high bioavailability.
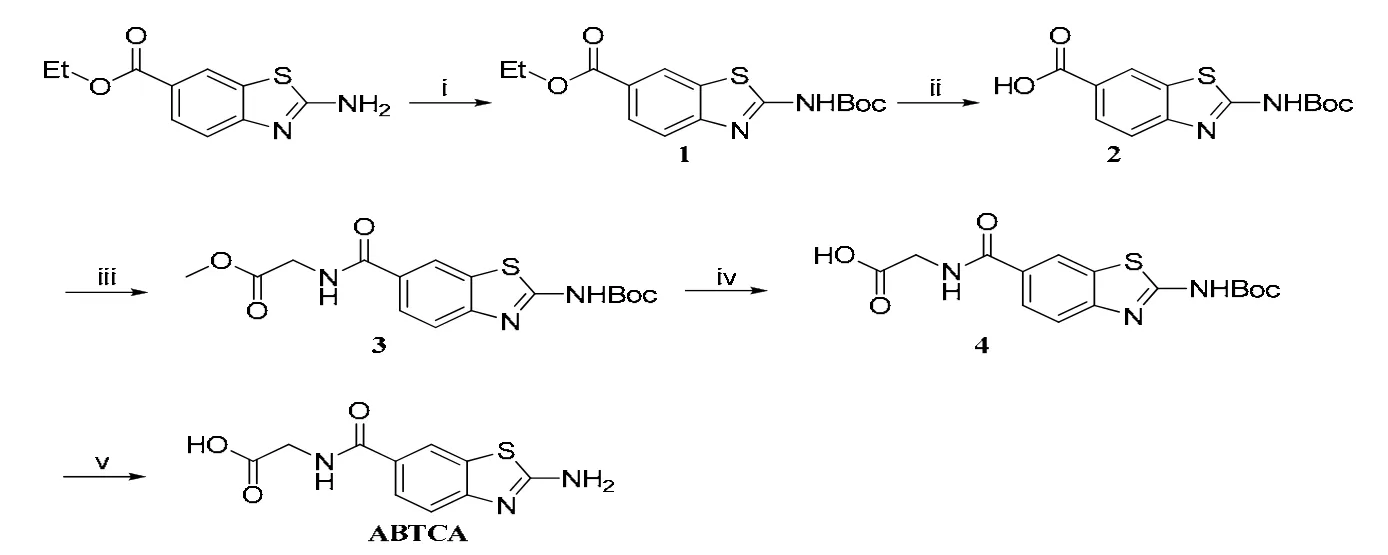
Scheme 2. Synthetic route for ABTCA. Reagent and condition: (i) 0.2 eq DMAP, 1.5 eq DIEA,1.2 eq (Boc)2O, THF, r.t.; (ii) 1 M NaOH in EtOH/THF/H2O (v:v = 1:1), 70 ℃;(iii) HBTU, DIEA, DMF, r.t.; (iv) LiOH, THF/H2O, r.t.; (v) TFA, DCM, r.t.
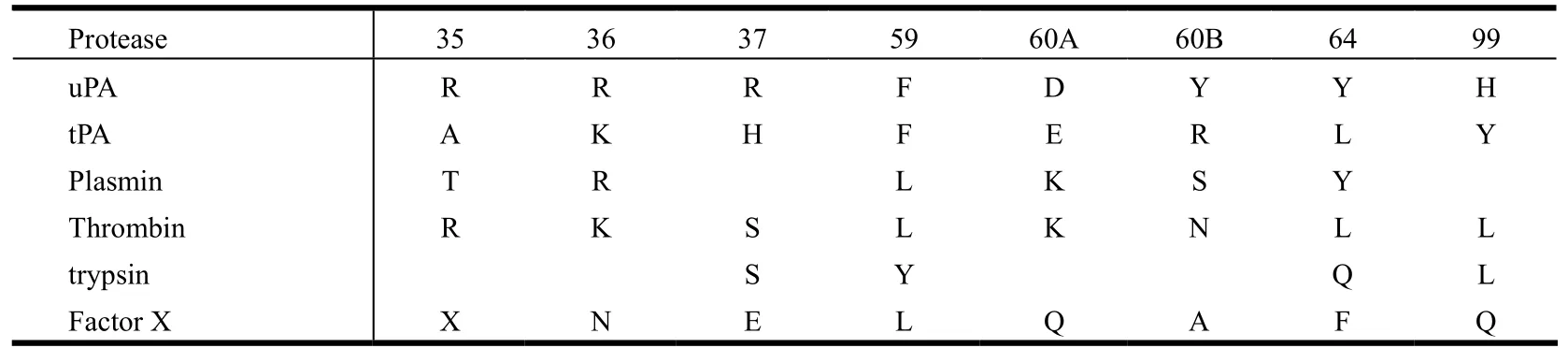
Table 3. Residues in 37- and 60-loops of uPA Compared with Other Selected Serine Proteases (All Residues Are in Chymotrypsin Numbering)
(1) Andreasen, P. A.; Kjoller, L.; Christensen, L.; Duffy, M. J. The urokinase-type plasminogen activator system in cancer metastasis: a review. Int. J.Cancer. 1997, 72, 1–22.
(2) Fazioli, F.; Blasi, F. Urokinase-type plasminogen activator and its receptor: new targets for anti-metastatic therapy? Trends Pharmacol. Sci. 1994, 15,25–29.
(3) Wysocki, A. B.; Kusakabe, A. O.; Chang, S.; Tuan, T. L. Temporal expression of urokinase plasminogen activator, plasminogen activator inhibitor and gelatinase–B in chronic wound fluid switches from a chronic to acute wound profile with progression to healing. Wound Repair Regen. 1999, 7,154–165.
(4) Rockway, T. W.; Nienaber, V.; Giranda, V. L. Inhibitors of the protease domain of urokinase-type plasminogen activator. Current Pharm. Design 2002,8, 2541–2558.
(5) Sperl, S.; Mueller, M. M.; Wilhelm, O. G.; Schmitt, M.; Magdolen, V.; Moroder, L. The uPA/uPA receptor system as a target for tumor therapy. Drug News Perspect. 2001, 14, 401–411.
(6) Setyono-Han, B.; Sturzebecher, J.; Schmalix, W. A.; Muehlenweg, B.; Sieuwerts, A. M.; Timmermans, M.; Magdolen, V.; Schmitt, M.; Klijn, J. G.;Foekens, J. A. Suppression of rat breast cancer metastasis and reduction of primary tumour growth by the small synthetic urokinase inhibitor WX-UK1. Thromb. Haemost. 2005, 93, 779–786.
(7) Menear, K. Progress towards the discovery of orally active thrombin inhibitors. Curr. Med. Chem. 1998, 5, 457–468.
(8) Albert, A.; Goldacre, R.; Phillips, J. The strength of heterocyclic bases. J. Chem. Soc. 1948, 2240–2249.
(9) Zhao, G. X.; Yuan, C.; Bian, C. B.; Jiang, L. G.; Ye, X. M.; Huang, Z. X.; Huang, M. D. Expression, purification and crystallization of urokinase catalytic domain in pichia pastoris. Prog. Biochem. Biophy. 2006, 33, 377–381.
(10) Fish, P. V.; Barber, C. G.; Brown, D. G.; Butt, R.; Collis, M. G.; Dickinson, R. P.; Henry, B. T.; Horne, V. A.; Huggins, J. P.; King, E.; O'Gara, M.;McCleverty, D.; McIntosh, F.; Phillips, C.; Webster, R. Selective urokinase-type plasminogen activator inhibitors. 4.1-(7-sulfonamidoisoquinolinyl)guanidines. J. Med. Chem. 2007, 50, 2341–2351.
(11) Leslie, A. G. W. Collaborative Computational Project No. 4. The CCP4 Suite∶ Programs for Protein Crystallography. Acta Crystallogr. D–Biol.Crystallogr. 1994, 50, 760–763.
(12) Brunger, A. T.; Adams, P. D.; Clore, G. M.; DeLano, W. L.; Gros, P.; Grosse-Kunstleve, R. W.; Jiang, J. S.; Kuszewski, J.; Nilges, M.; Pannu, N. S.;Read, R. J.; Rice, L. M.; Simonson, T.; Warren, G. L. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D–Biol. Crystallogr. 1998, 54, 905–921.
(13) Emsley, P.; Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr D–Biol Crystallogr. 2004, 60, 2126–2132.
(14) DeLano, W. L. The PyMOL molecular graphics system. DeLano Scientific, Palo Alto, CA, USA 2002.
(15) Wu, C.; Wei, J.; Gao, K.; Wang, Y. Dibenzothiazoles as novel amyloid–imaging agents. Bioorg. Med. Chem. 2007, 15, 2789–2796.
(16) Serdons, K.; Terwinghe, C.; Vermaelen, P.; Van Laere, K.; Kung, H.; Mortelmans, L.; Bormans, G.; Verbruggen, A. Synthesis and evaluation of 18F-labeled 2-phenylbenzothiazoles as positron emission tomography imaging agents for amyloid plaques in Alzheimer's disease. J. Med. Chem.2009, 52, 1428–1437.
(17) Malik, J. K.; Noolvi, M. N.; Manvi, F. V.; Nanjwade, B. K.; Patel, H. M.; Manjula, S. N.; Rao, M. C.; Barve, A. Synthesis and preliminary in-vitro cytotoxic activity of novel substituted diaryl-imidazo [2,1,b]-benzothiazole derivatives. Lett. Drug Des. Discov. 2011, 8, 717–724.
(18) Jiang, L. G.; Yu, H. Y.; Yuan, C.; Wang, J. D.; Chen, L. Q.; Edward, J. M.; Huang, Z. X.; Huang, M. D. Crystal structures of 2-aminobenzothiazole-based inhibitors in complexes with urokinase-type plasminogen activator. Chin. J. Struct. Chem. 2009, 28, 1427~1432.
(19) Nienaber, V.; Wang, J.; Davidson, D.; Henkin, J. Re-engineering of human urokinase provides a system for structure-based drug design at high resolution and reveals a novel structural subsite. J. Biol. Chem. 2000, 275, 7239–7248.
(20) Schweinitz, A.; Steinmetzer, T.; Banke, I. J.; Arlt, M. J.; Sturzebecher, A.; Schuster, O.; Geissler, A.; Giersiefen, H.; Zeslawska, E.; Jacob, U.; Kruger,A.; Sturzebecher, J. Design of novel and selective inhibitors of urokinase-type plasminogen activator with improved pharmacokinetic properties for use as antimetastatic agents. J. Biol. Chem. 2004, 279, 33613–33622.
(21) Zeslawska, E.; Jacob, U.; Schweinitz, A.; Coombs, G.; Bode, W.; Madison, E. Crystals of urokinase type plasminogen activator complexes reveal the binding mode of peptidomimetic inhibitors. J. Mol. Biol. 2003, 328, 109–118.
(22) Spraggon, G.; Phillips, C.; Nowak, U. K.; Ponting, C. P.; Saunders, D.; Dobson, C. M.; Stuart, D. I.; Jones, E. Y. The crystal structure of the catalytic domain of human urokinase-type plasminogen activator. Structure 1995, 3, 681–691.
(23) Hof, P.; Mayr, I.; Huber, R.; Korzus, E.; Potempa, J.; Travis, J.; Powers, J. C.; Bode, W. The 1.8 A crystal structure of human cathepsin G in complex with Suc-Val-Pro-PheP-(OPh)2: a Janus-faced proteinase with two opposite specificities. EMBO J. 1996, 15, 5481–5491.
(24) Katz, B. A.; Liu, B.; Barnes, M.; Springman, E. B. Crystal structure of recombinant human tissue kallikrein at 2.0 A resolution. Protein Sci. 1998, 7,875–885.
(25) Pike, A. C.; Brzozowski, A. M.; Roberts, S. M.; Olsen, O. H.; Persson, E. Structure of human factor VIIa and its implications for the triggering of blood coagulation. Proc. Natl. Acad. Sci. USA 1999, 96, 8925–8930.
(26) Renatus, M.; Engh, R. A.; Stubbs, M. T.; Huber, R.; Fischer, S.; Kohnert, U.; Bode, W. Lysine 156 promotes the anomalous proenzyme activity of tPA:X-ray crystal structure of single-chain human tPA. EMBO J. 1997, 16, 4797–4805.
(27) Syed Ibrahim, B.; Pattabhi, V. Trypsin inhibition by a peptide hormone: crystal structure of trypsin-vasopressin complex. J. Mol. Biol. 2005, 348,1191–1198.
(28) Katz, B. A.; Elrod, K.; Luong, C.; Rice, M. J.; Mackman, R. L.; Sprengeler, P. A.; Spencer, J.; Hataye, J.; Janc, J.; Link, J.; Litvak, J.; Rai, R.; Rice,K.; Sideris, S.; Verner, E.; Young, W. A novel serine protease inhibition motif involving a multi-centered short hydrogen bonding network at the active site. J. Mol. Biol. 2001, 307, 1451–1486.

- 结构化学的其它文章
- Synthesis and Crystal Structure of 5,5΄-Bis(4-hydroxylphenyl)diazo-dipyrromethane①
- A Novel Cu-based Metallosalan Complex:Synthesis, Structure and Chiral Sensor Study①
- Syntheses, Crystal Structures and Properties of Two Metal-organic Complexes Based on 5-(4-Pyridyl)-methoxyl Isophthalic Acid①
- DFT Study on the Electronic and Structural Properties of MoS6-/0 Clusters①
- Synthesis, Crystal Structure, and Biological Activity of 4-Chlorobenzaldehyde(2-trifluoromethylTrifluoromethyl-5,6,7,8-tetrahydrobenzo[4,5]- thieno[2,3-d]pyrimidin-4-yl)hydrazone Monohydrate①
- Synthesis and Crystal Structure of (Z)-N-(2-(diethylamino)ethyl)-7-(5-fluoro-2-oxoindolin-3-ylidene)-2-methyl-4,5,6,7-tetrahydro-1H-indole-3-carboxamide Methanol Solvate①