
三尖杉酯碱侧链酸的不对称合成及结构表征
2013-10-11 06:19孙默然曹其伟高凯歌
化学研究 2013年4期
孙默然,周 航,曹其伟,高凯歌,杨 华
(郑州大学 药学院,河南 郑州450001)
三尖杉酯碱(Harringtonine,HT)2是从海南粗榧属植物中提取的抗肿瘤药物,具有细胞毒、DNA合成抑制及染色体损伤等作用[1-2],能诱导白血病细胞HL60凋亡[3-4].目前,应用于临床的三尖杉酯碱的来源主要依赖于从三尖杉植物中提取,而它在植物体内的含量甚微.在三尖杉植物提取物中,无药理活性的三尖杉碱1的含量最高,约占总碱量的50%.因此,由提取的三尖杉碱1[2]和侧链酸半合成三尖杉酯碱2无疑可以变“废”为宝,提高资源综合利用率.

三尖杉酯碱侧链二酸及环状等效体的合成已有几例报道[5-7],其共同特点是以光学活性的苹果酸、柠檬酸或环氧化物作为手性源,通过烷基酮锂或格氏试剂的亲核加成反应引入烷基,由此构筑分子中关键的手性季碳.然而,烷基化过程需要强碱、低温及复杂的后处理过程,不利于放大生产.作者近期将从未用于构筑手性季碳的[2,3]-Meisenheimer重排反应,应用于三尖杉酯碱和高三尖杉酯碱侧链酸的合成[8].在路线设计上,重排前体中包含了除羟基外的分子骨架,手性叔醇是通过重排反应迁移而来,同时手性源L-氨基酸的手性得到了100%迁移.本文作者对路线多处步骤进行改进,使其操作更加简便,目前本实验室以5gL-苏氨酸为原料,在一周内即可得到7~9g环状等效体11.合成路线见图1.
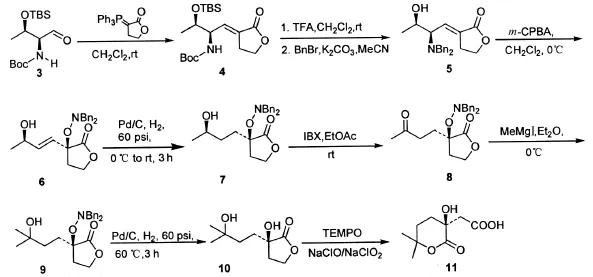
图1 目标化合物的合成路线Fig.1 Synthesis of the target compound
1 实验部分
1.1 仪器与试剂
WC-1型显微数字熔点仪;Nicolet IR200红外光谱仪(KBr压片);Bruker DPX-400核磁波谱仪(CDCl3为溶剂,TMS为内标);电喷雾四极杆飞行时间串联质谱仪(ESI-QTof).
化合物5按文献方法自制[8],其余所用试剂均为化学纯或分析纯,无水溶剂均按标准方法处理.
1.2 标题化合物的合成
1.2.1 化合物7的制备
称取化合物5(5.0g,14.40mmol),溶于50mL无水CH2Cl2,冰浴条件下搅拌10min后加入m-CPBA(75%含量)(3.66g,15.91mmol).30min后处理反应,过滤反应过程中生成的大量白色固体.收集滤液,二氯甲烷稀释,先后用10%NaOH (10mL×3)、饱和NaHCO3(10mL×2)、饱和食盐水(10mL×2)洗,无水MgSO4干燥,过滤.低于40℃水温条件下减压蒸馏,抽干至恒重,得到无色油状物6,无需纯化直接用于下步反应.
将上步反应所得油状物和10%Pd/C(湿重65.52%)(1.54g)加入250mL加氢瓶中,加入事先已冷却至0℃的无水甲醇120mL,置于加氢装置中.H2置换体系中的空气,反应自然升至室温反应3h.过滤反应液中的黑色固体,滤饼用大量甲醇冲洗,滤液减压浓缩抽干,柱层析(石油醚/乙酸乙酯,2/1)得到白色固体7(4.56g,86%).[α]20D= 40.3 (c= 1.84,EtOH);mp 176.2 ℃(PE/EtOAc);1H NMR(400MHz,CDCl3):δ=7.41~7.19(m,10H),4.40(d,J=12.8Hz,1H),4.20(dd,J=8.4Hz,5.6H2,1H),4.10~4.15(m,1H),3.90~3.81(m,2H),3.81~3.77(m,1H),3.62(d,J=13.6Hz,1H),1.97(brs,1H),2.06~1.84(m,1H),1.82~1.41(m,6H),1.18 (d,J= 2.4Hz,3H);13C NMR (100 MHz,CDCl3):δ=176.4,137.1,136.7,130.4,130.0,128.4,128.3,127.7,127.5,82.7,67.8,65.9,63.5,61.9,32.9,31.8,30.9,23.4;HRMS(ESI):m/z[M + Na]+calcd for C22H27NO4Na 392.182 4,found 392.182 5.
1.2.2 化合物8的制备
称取化合物7(2.01g,5.44mmol),IBX(3.05g,10.88mmol),加无水乙酸乙酯20mL溶解,加热回流3h后,过滤掉反应体系中的白色固体,滤液减压浓缩抽干,柱层析(乙酸乙酯/石油醚,3/1)得到白色固体8(1.78g,89%).[α]20D=76.5(c=1.21,EtOH);mp 99.3℃ (PE/EtOAc);IR (KBr,thin film)νmax:3 029,2 963,1 765,1 713cm-1;1H NMR(400MHz,CDCl3):δ=7.43~7.16(m,10H),4.51(d,J= 12.8Hz,1H),4.25~4.18(m,1H),4.10~4.02(m,1H),3.87(dd,J=7.6Hz,13.2,1H),3.58(d,J=13.6Hz,1H),2.76~2.64(m,1H),2.64~2.53(m,1H),2.81(s,3H),2.08~1.97(m,1H),1.95~1.80(m,1H),1.51~1.42(m,1H);13C NMR (100MHz,CDCl3):δ= 170.1,138.3,133.9,131.7,128.8,128.7,127.6,65.6,65.5,65.0,54.2,25.9,19.3;HRMS(ESI):m/z[M +Na]+calcd for C22H25NO4Na 390.173 8,found 390.1 73 5.
1.2.3 化合物9的制备
取25mL两口圆底烧瓶,放置Mg屑(176mg,7.78mmol)并用N2置换空气,向反应瓶加入5mL无水Et2O、含 MeI(0.49mL,7.78mmol)的无水Et2O溶液3mL,搅拌至 Mg屑完全消失,即制成甲基格氏试剂.
另在100mL两口圆底烧瓶中加入化合物8(1.43g,3.89mmol),加入无水Et2O与无水甲苯的混合溶剂60mL(Et2O∶无水甲苯=2∶1),用N2置换空气.将上步新制备的甲基格氏试剂逐滴缓慢地滴加入反应体系,继续搅拌30min后,加入饱和NH4Cl溶液淬灭反应,乙酸乙酯(40mL×3)萃取,合并有机相,用饱和食盐水洗两次、无水MgSO4干燥、过滤、浓缩,柱层析(乙酸乙酯/石油醚,3/1)得到无色油状物9(1.17g,79%).[α]20D=50.8(c=1.54,EtOH);mp 114.7℃(PE/EtOAc);IR (KBr,thin film)νmax:3 535,3 463,1 759,1 492cm-1;1H NMR(400MHz,CDCl3):7.38~7.20(m,10H),4.42(d,J=12.8Hz,1H),4.26(dd,J=8.0Hz,15.2H2,1H),4.11~4.04(m,1H),3.85(q,J=6.4Hz,2H),3.61(d,J=13.2Hz,1H),1.78~1.60(m,5H),1.43~1.35(m,2H),1.23(d,J=6.0Hz,3H);13C NMR(100MHz,CDCl3):176.6,137.1,136.6,130.5,130.2,129.9,128.4,127.7,127.5,82.8,70.4,66.0,63.5,61.9,36.9,30.7,30.4,29.4,29.1;HRMS(ESI):m/z[M + Na]+calcd for C23H29NNaO4406.201 5,found 406.201 9.
1.2.4 化合物10的制备
称取化合物9(4.30g,12.20mmol),10%Pd/C(湿重65.52%)(1.25g)于250mL加氢瓶中,加入无水甲醇150mL,置于加氢装置上,H2置换体系中的空气,60℃反应3h后,过滤掉反应液中的固体,甲醇溶剂浓缩至干,柱层析(石油醚/乙酸乙酯,1/2)得到白色固体10(1.85g,81%).[α]20D=18.7(c=1.09,EtOH);mp 98.4℃(PE/EtOAc);IR(KBr,thin film)νmax:3 416,3 258,1 752,1 404cm-1;1H NMR(400MHz,CDCl3):δ=4.93(s,1H),4.43~4.38(m,1H),4.25~4.18(m,1H),2.55(brs,1H),2.44~2.35(m,1H),2.28~2.19(m,1H),1.98~1.83(m,2H),1.70(t,J=6.8Hz,2H),1.27(d,J=5.2Hz,6H);13C NMR(100MHz,CDCl3):δ=19.2,74.2,70.76,65.3,36.9,35.8,31.2,30.2,28.3;HRMS(ESI):m/z[M + Na]+calcd for C9H16NaO4211.095 3,found 211.095 1.
1.2.5 化合物11的制备
合成方法和波谱数据参见文献报道[8].
2 结果与讨论
2.1 Meisenheimer重排反应前体5的合成
以L-苏氨酸为原料,经五步常规反应得到N-Boc氨基醛3[9],未经纯化的化合物3与丁酸内酯魏悌息试剂在无水CH2Cl2中室温反应2h即可顺利转化成化合物4,且该反应中的副产物三苯氧膦与产物的Rf值相差较大,并与产物不粘连,很容易分离,避免了一般Wittig反应三苯氧膦难以除去的难题,大大减少了工作量.随后的脱保护反应,意外地发现三氟乙酸不仅仅脱去Boc保护基,还将羟基保护基TBS也脱除了,文献检索未见三氟乙酸条件下脱去TBS保护基的报道.Meisenheimer重排反应需在氮上双苄基取代条件下进行.脱保护产物蒸除溶剂和未反应的三氟乙酸,直接加入3倍量的K2CO3和2.2倍量的苄溴,加热回流,成功转化为N-双苄基保护氮氧化物5.
2.2 化合物8的合成
化合物5在无水CH2Cl2中,加入1.1倍量的m-CPBA,0℃条件反应30min,得到单一产物6.作者近期文献报道[8],为了得到多用途的手性构筑砌块,化合物6经锌-醋酸还原法切断N-O键,保留了烯烃双键.但此法后处理需柱层析分离副产物二苄胺,不利于放大生产.我们试图用催化氢化法一锅内还原N-O键和双键,但TLC监测到多个产物点.原因可能是还原产物具有γ-羟基酮结构片段,发生分子内亲核加成反应,生成五元环状半缩酮[10].因此,我们改变策略,将催化氢化条件设在冰浴到室温之间,得到N-O键被保留的化合物7.由此双苄基氨可以充当羟基保护基,避免发生分子内成环反应.化合物6与7极性差别细微,二者Rf值近乎相同,且均有紫外吸收,只能通过磷钼酸显色快慢与颜色的差别做出区分.化合物7经IBX氧化转化为酮8.相比于MnO2氧化反应[8],IBX氧化反应的后处理更易操作,将反应液过滤,除去不溶于乙酸乙酯的未反应完的IBX和副产物IBN,并用大量乙酸乙酯洗涤滤饼,滤液旋蒸至干即可得到化合物8.
2.3 三尖杉酯碱侧链酸环等价体11的合成
化合物8在无水乙醚中的溶解度差,因此选用无水乙醚/无水甲苯体积比2∶1混合溶剂增加其溶解性.文献方法甲基溴化镁对酮进行亲核加成,由于溴甲烷是气体,反应需要在-78℃下进行.此路线以碘甲烷代替溴甲烷制备甲基格氏试剂,在温和的反应条件下(0℃)得到化合物9.再经60℃催化氢化断开N-O键,得到丁内酯10.δ-羟基羧酸在酸性条件下易发生内酯化反应,TEMPO氧化[11]高收率得到三尖杉碱侧链二酸等效体δ-内酯11.化合物11的1H NMR、13C NMR、熔点和比旋光度均与文献报道[5]相符.
3 结论
综上所述,本文报道了以N-Boc苏氨醛3为起始原料,经九步反应,总收率30%,不对称合成三尖杉酯碱侧链酸的新路线.合成天然产物过程中使用的原料价廉易得,实验条件温和,操作简单,具备放大生产的条件.
[1]潘震昆,韩 锐,王永潮.三尖杉酯碱对白血病L-1210细胞杀伤动力学研究:Ⅰ.放射自显影观察[J].生物化学与生物物理学报,1980,12(1):13-20.
[2]潘维林,王端顺,王永潮.用早熟染色体凝集(PCC)法和克隆培养法研究三尖杉酯碱对细胞杀伤动力学[J].中华肿瘤杂志,1983,5(4):256-259.
[3]孟凡宏,何琪杨,池旭生,等.人白血病HL60细胞的分化状态对细胞凋亡的影响[J].药学学报,1997,32(7):496-501.
[4]李 程,曹丽芝,万恂恂,等.三尖杉酯碱对HL60细胞凋亡及原癌基因表达的影响[J].中国病理生理杂志,2001,17(2):107-119.
[5]SERRY A A,EL B,HOLGER B,et al.Enantioselective synthesis ofα-alkylmalates as the pharmacophoric group of several natural alkaloids and glycosides[J].Eur J Org Chem,2005:2965-2972.
[6]RACHAEL A A,ANDREW T R,ADAM J S.Synthesis of the ester side chains of some potently antileukemic harringtonia alkaloids from chiral citrates[J].Chem Comm,2006,30:3243-3245.
[7]FAROUK B,SEBASTIEN T,JOELLE P V,et al.Synthesis of optically active monoacid side-chains of cephalotaxus alkaloids[J].Eur J Org Chem,2009(3):437-443.
[8]YANG H,SUN M R,ZHAO S G,et al.Construction of chiral tertiary alcohol stereocentersviathe[2,3]-meisenheimer rearrangement:enantioselective synthesis of the side-chain acids of homoharringtonine and harringtonine[J].J Org Chem,2013,78(2):339-346.
[9]KUTSUMURA N,NISHIYAMA S J.Synthetic studies of N-demethylossamine and elaboration of its glycosylation[J].J Carbohydrate Chem,2006,25(5):377-385.
[10]CHRISTIANE B,HILTRUD H,HANS-ULRICH R.Diastereoselective syntheses of highly substituted methyl tetrahydrofuran-3-carboxylates by reactions of y-lactols with silylated nucleophiles[J].J Org Chem,1988,53(11):2450-2456.
[11]FRIEDRICH P,DARLEY D J,GOLDING B T,et al.Complete stereochemistry of the enzymatic dehydration of 4-hydroxybutyryl coenzyme A to crotonyl coenzyme A [J].Angew Chem Int Ed,2008,47(17):3254-3257.
猜你喜欢
分子催化(2022年1期)2022-11-02
石油化工自动化(2020年1期)2020-03-05
通信技术(2019年8期)2019-09-03
吉林农业(2019年6期)2019-06-11
国际呼吸杂志(2019年4期)2019-03-12
中成药(2017年10期)2017-11-16
中成药(2017年4期)2017-05-17
国外医药(抗生素分册)(2016年4期)2016-07-12
国外医药(抗生素分册)(2016年2期)2016-07-12
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01
