
1-甲基胸腺嘧啶A-带结构动力学
2013-09-21 09:00李明娟刘明霞郑旭明
物理化学学报 2013年5期
李明娟 刘明霞 郑旭明
(浙江理工大学化学系,先进纺织材料与加工技术教育部重点实验室,生态染整技术教育部工程研究中心,杭州310018)
1 引言
胸腺嘧啶(T)是DNA中最容易发生光二聚的碱基,1,2其激发态衰变动力学及其甲基化和水中氢键的影响一直是研究热点.3-8Perun等9的研究表明,胸腺嘧啶的最低单重态Sπ和基态S0间的圆锥交叉过程是一个无能垒的超快非辐射衰变过程.飞秒光谱对核酸碱基的研究揭示了胸腺嘧啶与胸苷的最初单重激发态(SFC)无辐射衰变主要经过两条内转换(IC)途径:一条是通过Sπ/S0势能面的圆锥交叉由最初单重激发态衰变到基态(S0);10-12另一条是通过Sπ/Sn势能面的圆锥交叉由最初单重激发态衰变到1nπ*态(暗态).13-15这两条IC途径都发生在亚皮秒时间内,而1nπ*态可以进一步衰变到基态(S0)或进一步产生激发三重态(T1).16,17Zgierski等18阐明嘧啶碱基处在非平面性激发态结构时有利于促进超快非辐射衰退过程.理论计算还发现,由面外振动模促成的Sπ/Sn振动电子耦合是核酸碱基Sπ→Sn无辐射跃迁的重要途径.2,19,20共振拉曼光谱强度分析技术研究胸腺嘧啶等在质子与非质子性溶剂中的激发态短时动力学已有报道.21-24在水中,N1-糖基化有效隔离了胸腺嘧啶的振动,而N1-甲基化改变了胸腺嘧啶的均质和非均质线宽.23胸腺嘧啶在水中的结构动力学与在乙腈溶剂中的明显不同,说明溶剂对激发态势能面有重要的调控作用.24本文利用共振拉曼光谱结合密度泛函理论研究1-甲基胸腺嘧啶(MT)激发态结构行为,考察N1甲基化对胸腺嘧啶激发态结构动力学的影响及其溶剂调控规律.
2 实验部分
2.1 试剂和仪器
1-甲基胸腺嘧啶(98.0%,Sigma-Aldrich公司),乙腈(光谱纯,99.9%,TEDIA公司),甲醇(分析纯,≥99.5%,天津市永大化学试剂有限公司),水(分析纯,99.0%,自制).
紫外分光光度计(UV-2501PC,岛津公司,日本),傅里叶变换红外光谱仪(Nicolet Avatar370,美国),傅里叶变换拉曼光谱仪(Nicolet Raman 960,美国),共振拉曼光谱仪(自制).
2.2 实验方法
3 理论计算
电子基态几何结构和简正振动频率用密度泛函理论在B3LYP/6-311++G(d,p)水平计算获得;电子跃迁能和振子强度在B3LYP-TD/6-311++G(d,p)水平计算得到.本文所有量子化学计算均由Gaussian 09W程序包29完成.
4 结果和讨论
4.1 电子光谱

图1 MT的几何结构示意图Fig.1 Schematic diagram of the geometry structure of MT

图2 MT在乙腈、甲醇、水溶剂中的紫外吸收光谱Fig.2 UV absorption spectra of MT in acetonitrile,methanol,and water solvents
图1 示出由B3LYP/6-311++G(d,p)计算水平下优化获得的MT几何结构示意图.MT的对称性属于Cs点群,与文献6,24报道的胸腺嘧啶的对称性一致.图2是MT在乙腈、甲醇、水溶剂中的紫外吸收光谱,箭头所示的数值为共振拉曼光谱实验所用的激发波长.由图2可见,MT的紫外光谱大约在270和208 nm处有两个吸收带(分别称为A带和B带).溶剂对紫外光谱的摩尔消光系数和最大吸收波长影响不大,表明MT的A带和B带吸收的电子跃迁性质与溶剂的质子性或非质子性关系不大.依据Lambert-Beer定律:A=ε·b·c,实验测得的MT在乙腈、甲醇、水中的摩尔消光系数εmax分别为8.55×103、8.78×103、8.69×103L·mol-1·cm-1.
表1列出由B3LYP-TD/6-311++G(d,p)计算水平和极化连续介质模型(PCM)计算获得的MT电子跃迁能(ΔE)和振子强度(f).结果表明,计算获得的260 nm(f=0.2367)和207 nm(f=0.2062)吸收带与实验值270 nm(f=0.2061)和208 nm(f=0.2954)吻合良好.为了更清楚地理解MT激发态的主要跃迁轨道及电子云分布,图3示出MT的主要跃迁轨道.轨道37、35、38和40分别为πH、πH-2、πL*和π*L+2分子轨道;轨道36和34是两个非键轨道nH-1和nH-3;轨道39是弥散轨道Ryd1,弥散轨道Ryd1的电子振幅主要在CH3(C7)和C6―H12这4个氢原子上.因此,表1中的A带吸收可指认为πH→πL*/πH-2→π*L+2跃迁,而 B 带吸收可指认为πH→π*L+2/πH-2→πL*跃迁.与胸腺嘧啶的跃迁轨道比较后得知,MT中甲基与嘧啶环的超共轭效应导致了A带吸收的明显红移.
4.2 振动光谱指认
MT的振动光谱指认已有理论计算研究报道.30,31为了开展共振拉曼光谱指认和激发态结构动力学研究,我们结合振动光谱实验,完善了MT的振动光谱指认.由B3LYP/6-311++G(d,p)计算获得拉曼光谱和实验测得的FT-IR和FT-Raman光谱示于图4(A)和列于表2.计算拉曼光谱共有48个振动模.在0-1500 cm-1光谱区域内,MT傅里叶拉曼光谱出现25个振动带,其中A'不可约表示占20个,A''不可约表示占5个,它们均为红外和拉曼活性模.

表1 在PCM溶剂模型下由B3LYP-TD/6-311++G(d,p)计算获得的MT电子跃迁能(ΔE),轨道和振子强度(f)Table 1 Electronic transition energies(ΔE),the corresponding orbitals and the oscillator strengths(f)for MT computed by B3LYP-TD/6-311++G(d,p)and polarization continuum moldel(PCM)

图3 表1中MT电子跃迁相关的分子轨道Fig.3 Molecular orbitals associated with the electronic transitions of MT as listed in Table 1
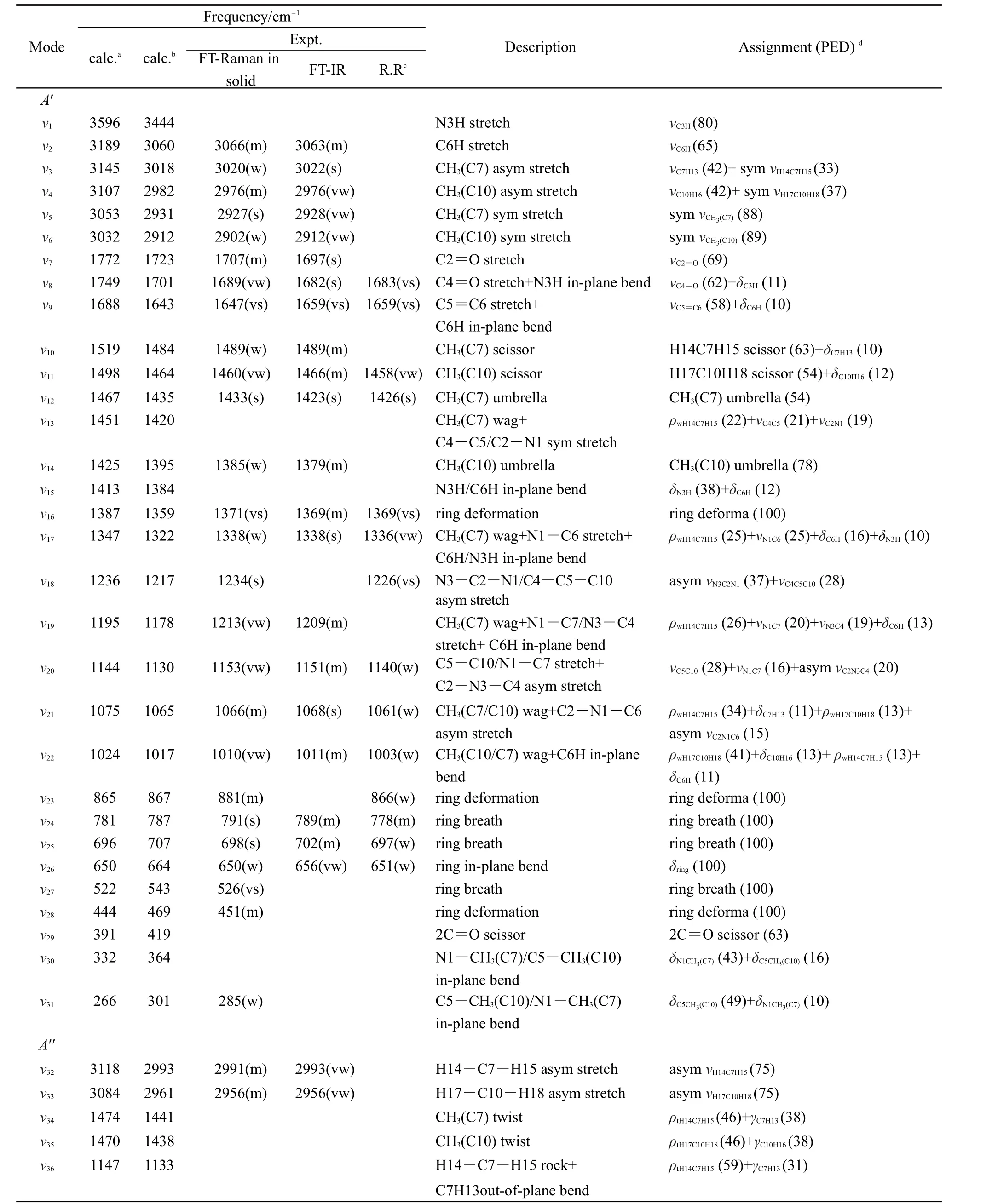
表2 MT在B3LYP/6-311++G(d,p)计算水平下的计算频率和实验傅里叶变换红外光谱与傅里叶变换拉曼光谱观察到的振动Table 2 Vibrational frequencies and assignments of MT computed by B3LYP/6-311++G(d,p)and observed by experimental FT-IR and FT-Raman spectra

continued Table 2
图4(B)示出不同溶剂的266 nm共振拉曼光谱与傅里叶拉曼光谱和计算拉曼光谱的对比图.表2和图4(A)中1500-1800 cm-1光谱区域的三个振动模的指认得到了共振拉曼光谱的确认.计算拉曼频率ν9、ν8和ν7分别位于1661、1720和1743 cm-1.傅里叶拉曼光谱在1647 cm-1处是一个强峰,在1689和1707 cm-1处是两个弱峰.由于分辨率的局限,傅里叶红外光谱展示一个大吸收峰,其主峰位于1655 cm-1,肩峰位于1697 cm-1.在乙腈中266 nm共振拉曼光谱展示1659和1683 cm-1两个强峰,它们分别对应于傅里叶变换拉曼光谱中的1647和1689 cm-1峰和计算拉曼光谱中的1661和1720 cm-1峰,因此分别被指认为ν9和ν8.而傅里叶拉曼光谱中的1707 cm-1峰被指认为ν7.根据表2的指认结果对计算频率与实验频率数据进行线性回归分析,得出标准偏差(SD)为14 cm-1,说明计算拉曼频率值与实验振动频率总体吻合较好,指认结果合理.
4.3 共振拉曼光谱和结构动力学
图5示出MT在乙腈和水溶剂中282.4、273.9、266.0、228.7和208.8 nm共振拉曼光谱在500-1800 cm-1光谱区域的振动基频信息,其中282.4、273.9和266.0 nm共振拉曼光谱涵盖A吸收带,其强度模式主要由 S1(A')或1πHπL*电子激发态势能面决定,而228.7和208.8 nm共振拉曼光谱涵盖B吸收带,其强度模式由S4(A')或1πHπ*L+2电子激发态势能面决定.由图5可见,在乙腈溶剂中,266.0、273.9、282.4 nm共振拉曼谱图彼此十分类似,有14个活性振动模(ν8,ν9,ν11,ν12,ν16,ν17,ν18,ν20,ν21,v22,ν23,ν24,ν25,ν26),它们的主要特征为:C4=O9伸缩振动ν8(1683 cm-1),C5=C6伸缩振动ν9(1659 cm-1),H17C10H18剪式振动+C10H16面内弯曲振动ν11(1458 cm-1),CH3(C7)伞式振 动 ν12(1426 cm-1),环 变 形 振 动 ν16(1369 cm-1),H14C7H15面外摇摆振动+N1C6伸缩振动+C6H/N3H面内弯曲振动ν17(1336 cm-1),N3C2N1反对称伸缩+C4C5C10反对称伸缩振动ν18(1226 cm-1),C5C10伸缩+N1C7伸缩+C2N3C4反对称伸缩ν20(1140 cm-1),CH3(C7/C10)面外摇摆振动+C2N1C6不对称伸缩振动ν21(1061 cm-1),CH3(C10/C7)面外摇摆振动+C6H12面内弯曲振动ν22(1003 cm-1),环变形振动ν23(866 cm-1),环呼吸振动v24(778 cm-1),环呼吸振动ν25(697 cm-1),环面内弯曲振动ν26(651 cm-1).

图4 (A)MT的傅里叶变换红外、傅里叶变换拉曼和计算拉曼光谱的比较;(B)傅里叶变换拉曼、计算拉曼和乙腈、甲醇及水中MT的266 nm共振拉曼光谱的比较Fig.4 (A)Comparison of FT-IR,FT-Raman and computed Raman spectra of MT;(B)comparison of FT-Raman,computed Raman,and the 266 nm resonance Raman spectra of MT in acetonitrile,methanol,and water solvents The spectra were indicated with the frequency values and tentative vibrational assignments for larger Raman features.
208.8 和228.7 nm共振拉曼谱共有11个活性振动模(ν8,ν9,ν11,ν16,ν17,ν18,ν20,ν21,ν23,ν24,ν25).显然,A带共振拉曼光谱的强度模式与B带共振拉曼光谱的强度模式存在很大差别,说明它们的激发态结构动力学明显不同.ν11振动模的基频、泛频及其与其它振动模的合频占据了B带共振拉曼光谱强度的绝大部分,但在A带共振拉曼光谱中该振动模的强度极弱,这说明B吸收带预共振增强效应对A带共振拉曼强度模式的贡献可以忽略,因此266.0、273.9、282.4 nm共振拉曼光谱的强度模式主要反映A带吸收的结构动力学,而208.8、228.7 nm拉曼光谱振动模主要反映B带吸收时的动态结构.
由图5可见,水中A带共振拉曼光谱最强振动模为ν9、ν16、ν18,中等强度振动模为ν12、ν20、ν21、ν22、ν23、ν24和ν25.这说明,MT的结构动力学主要沿着C5=C6伸缩、环变形振动、N3C2N1/C4C5C10反对称伸缩振动等反应坐标位移,多维性特征明显.这表明,在光化学反应的初期,光激发后的可用能被分散到多个振动模上.A带共振拉曼光谱中ν9振动模具有最大强度,这与A带吸收的主要电子跃迁πH→πL*特征相吻合(详见图3中轨道37、38),因为πH→πL*跃迁大大减弱了C5=C6键的键级.与A带共振拉曼光谱不同,B带共振拉曼光谱强度的绝大多数被ν11振动模占据,表明其短时动力学主要沿着H17C10H18剪式振动和C10H16面内弯曲振动反应坐标展开.进一步比较MT与T在乙腈和水溶剂中的A带共振拉曼光谱后发现,MT比T多了ν22(CH3(C10/C7)面外摇摆振动+C6H12面内弯曲振动)、ν21(CH3(C7/C10)面外摇摆振动+C2N1C6不对称伸缩振动)、ν17(H14C7H15面外摇摆振动+N1C6伸缩振动+C6H/N3H面内弯曲振动)、ν12(CH3(C7)伞式振动)等4个振动模,它们均与1位甲基取代有关,这说明1位甲基取代并参与嘧啶环的共轭作用,对胸腺嘧啶的激发态振动重组能产生明显影响.
为了考察激发态反应坐标的位移量,图6示出乙腈和水中266.0 nm共振拉曼光谱中ν8、ν9及其倍频峰的强度.由图6可见,在乙腈中C4=O9伸缩振动ν8(1683 cm-1)的基频和倍频2ν8具有很大强度,而它们在水中变得很弱.这说明,MT的C4=O9伸缩振动模在乙腈中是活性的,而在水中为非活性或活性很弱.这一现象与胸腺嘧啶激发态动力学受溶剂影响规律一致.24因此,N1甲基化不改变质子性和非质子性溶剂对胸腺嘧啶的C4=O9反应坐标的调控规律.

图5 MT在乙腈(a)和水(b)溶剂中的共振拉曼光谱Fig.5 Resonance Raman spectra of MT in acetonitrile(a)and water(b)solvents

图6 266.0 nm激发波长下MT在水(a)与乙腈(b)溶剂中共振拉曼光谱指认Fig.6 Expanded view of the 266.0 nm resonance Raman spectra of MT in water(a)and acetonitrile(b)solvents
目前已有的理论计算和共振拉曼实验研究结果24表明,胸腺嘧啶在乙腈溶剂中存在Franck-Condon区域1ππ*/1nπ*势能面锥形交叉,而在水中则没有.在胸腺嘧啶中C4=O9伸缩振动的激活与失活被认为与1ππ*/1nπ*势能面锥形交叉直接相关.因此,根据水和乙腈中MT与胸腺嘧啶短时结构动力学的相似性,本文认为在Franck-Condon区域MT也存在类似的1ππ*/1nπ*势能面锥形交叉.这时,被激发到1ππ*激发态的MT分子,其量子波包的一部分沿Sp势能面衰变,而另一部分则通过1ππ*/1nπ*势能面锥形交叉衰变到1nπ*态.而在水中,由于氢键作用改变了1nπ*势能面的能量,使1ππ*/1nπ*势能面锥形交叉不能发生在Franck-Condon区域.因此,由于1nπ*态(禁阻跃迁,暗态)失去了与1ππ*(允许跃迁,明态)发生强烈振动电子耦合作用的条件,与1nπ*激发态相关的动态活性模ν8便失去了从1ππ*电子跃迁中借到强度的机会,使得在水中共振拉曼光谱中的C4=O9伸缩振动模的基频和泛频消失或大大减弱,在动力学上这意味额外的1nπ*通道消失或严重被削弱.32
5 结论
采用共振拉曼光谱技术结合密度泛函理论方法研究了1-甲基胸腺嘧啶的电子激发和Franck-Condon区域结构动力学,得出如下结论:
(2)在乙腈溶剂中,MT的A带和B带共振拉曼谱被分别指认为14个和11个活性振动模,其结构动力学主要沿着C5=C6和C4=O9伸缩振动和环变形振动坐标展开.水的氢键作用严重削弱了C4=O9伸缩振动坐标在MT结构动力学中的作用.
(3)与胸腺嘧啶相比,N1甲基化导致甲基与嘧啶环产生超共轭效应,引起A带吸收的明显红移,但不改变质子性和非质子性溶剂对胸腺嘧啶的C4=O9反应坐标的调控规律.
(1) Beukers,R.Biochemical Biophysical Acta 1960,41,550.doi:10.1016/0006-3002(60)90063-9
(2) Crespo-Hernández,C.E.;Cohen,B.;Hare,P.M.;Kohler,B.Chem.Rev.2004,104,1977.doi:10.1021/cr0206770
(3)Wetmore,S.D.;Boyd,R.J.Phys.Chem.B 1998,102,5369.doi:10.1021/jp9809078
(4) Etinski,M.;Marian,C.M.Phys.Chem.Chem.Phys.2010,12,4915.doi:10.1039/b925677f
(5) Busker,M.;Nispel,M.;Häber,T.;Kleinermanns,K.;Etinski,M.;Fleig,T.ChemPhysChem 2008,9,1570.doi:10.1002/cphc.v9:11
(6) Etinski,M.;Fleig,T.;Marian,C.M.J.Phys.Chem.A 2009,113,11809.doi:10.1021/jp902944a
(7) Kunitski,M.;Nosenko,Y.;Brutschy,B.ChemPhysChem 2011,12,2024.doi:10.1002/cphc.201000985
(8) Nosenko,Y.;Kunitski,M.;Brutschy,B.J.Phys.Chem.A 2011,115,9429.doi:10.1021/jp111373t
(9) Perun,S.;Sobolewski,A.L.;Domcke,W.J.Phys.Chem.A 2006,110,13238.doi:10.1021/jp0633897
(10) Schreier,W.J.;Schrader,T.E.;Koller,F.O.;Gilch,P.;Crespo-Hernández,C.E.;Swaminathan,V.N.;Carel,T.;Zinth,W.;Kohler,B.Science 2007,315,625.doi:10.1126/science.1135428
(11) Pecourt,J.M.L.;Peon,J.;Kohler,B.J.Am.Chem.Soc.2001,123,10370.doi:10.1021/ja0161453
(12) Gustavsson,T.;Sharonov,A.;Markovitsi,D.Chem.Phys.Lett.2002,351,195.doi:10.1016/S0009-2614(01)01375-6
(13)Hare,P.M.;Crespo-Hernández,C.E.;Kohler,B.Proc.Natl.Acad.Sci.U.S.A.2007,104,435.doi:10.1073/pnas.0608055104
(14) Hare,P.M.;Crespo-Hernández,C.E.;Kohler,B.J.Phys.Chem.B 2006,110,18641.doi:10.1021/jp064714t
(15)Kwok,W.M.;Ma,C.;Phillips,D.L.J.Am.Chem.Soc.2008,130,5131.doi:10.1021/ja077831q
(16) Marguet,S.;Markovitsi,D.J.Am.Chem.Soc.2005,127,5780.doi:10.1021/ja050648h
(17) Salet,C.;Bensasson,R.;Becker,R.S.Photochem.Photobiol.1979,30,325.doi:10.1111/php.1979.30.issue-3
(18) Zgierski,M.Z.;Patchkovskii,S.;Lim,E.C.J.Chem.Phys.2005,123,81101.doi:10.1063/1.2031207
(19) Merchán,M.;Serrano-Andrés,L.;Robb,M.A.;Blancafort,L.J.Am.Chem.Soc.2005,127,1820.doi:10.1021/ja044371h
(20) Blancafort,L.;Robb,M.A.J.Phys.Chem.A 2004,108,10609.doi:10.1021/jp045985b
(21)Yarasi,S.;Brost,P.;Loppnow,G.R.J.Phys.Chem.A 2007,111,5130.doi:10.1021/jp071443t
(22)Yarasi,S.;Ng,S.;Loppnow,G.R.J.Phys.Chem.B 2009,113,14336.
(23) Billinghurst,B.E.;Oladepo,S.A.;Loppnow,G.R.J.Phys.Chem.B 2012,116,10496.
(24)Zhu,X.M.;Wang,H.G.;Zheng,X.M.;Phillips,D.L.J.Phys.Chem.B 2008,112,15828.doi:10.1021/jp806248b
(25)Weng,K.F.;Wang,H.G.;Zhu,X.M.;Zheng,X.M.Acta Phys.-Chim.Sin.2009,25,1799.[翁克凤,王惠钢,祝新明,郑旭明.物理化学学报,2009,25,1799.]doi:10.3866/PKU.WHXB20090825
(26)Liu,C.;Du,R.;Zhao,Y.Y.;Wang,H.G.;Zheng,X.M.Acta Phys.-Chim.Sin.2011,27,17.[刘 崇,杜 蕊,赵彦英,王惠钢,郑旭明.物理化学学报,2011,27,17.]doi:10.3866/PKU.WHXB20110132
(27)Xu,Z.P.;Zhao,Y.Y.;Wang,H.G.;Zheng,X.M.Acta Phys.-Chim.Sin.2012,28,65.[许宗平,赵彦英,王惠钢,郑旭明.物理化学学报,2012,28,65.]doi:10.3866/PKU.WHXB20122865
(28) Myer,A.B.;Li,B.;Ci,X.J.Chem.Phys.1988,89,1876.doi:10.1063/1.455135
(29) Frisch,M.J.;Treucks,G.W.;Schlegel,H.B.;et al.Gaussian 09[M];Gaussian Inc.:Wallingford,CT,2009.
(30) Lagant,P.;Vergoten,G.;Peticolas,W.L.J.Raman Spectrosc.1999,30,1001.
(31)Morzyk-Ociepa,B.;Nowak,M.J.;Michalska,D.Spectrochimica Acta Part A 2004,60,2113.doi:10.1016/j.saa.2003.11.009
(32) Santoro,F.;Barone,V.;Gustavsson,T.;Improta,R.J.Am.Chem.Soc.2006,128,16312.doi:10.1021/ja0657861
猜你喜欢
昆明医科大学学报(2021年8期)2021-08-13
汕头大学学报(自然科学版)(2020年4期)2020-12-14
农药科学与管理(2019年6期)2019-11-23
武警医学(2018年10期)2018-11-06
中国粮油学报(2018年12期)2018-03-19
当代化工研究(2016年6期)2016-03-20
原子与分子物理学报(2015年3期)2015-11-24
读写算·教研版(2014年12期)2014-09-01
原子与分子物理学报(2014年1期)2014-03-20
原子能科学技术(2014年3期)2014-02-28
