
基于细菌群感效应人工构建分子开关
2013-09-04 08:35张志伟吴胜
生物工程学报 2013年9期
张志伟,吴胜
1 中国科学院微生物研究所 微生物资源前期开发国家重点实验室,北京 100101
2 中国科学院大学,北京 100049
在生物体内,一个多步骤的生物合成途径中往往存在一个或几个关键的节点受到多重水平的调节从而精确地控制目标产物的合成[1-3]。目前,对生物合成途径的改造,常规的做法是增加代谢途径中受调蛋白基因的拷贝数,过量表达目标蛋白。但这种对代谢途径的改造方法很多时候并不奏效,原因可能是目标蛋白的过量表达,往往会带来非常严重的负面效应。主要表现为:由于常规的蛋白质表达是一种不完善的受控表达,只能启动,不能终止,表达的蛋白质大部分在体内形成包涵体,不仅是对生物体内能量和物质的巨大浪费,而且造成细胞严重的生理负担。针对上述问题,我们提出基于细菌群感效应的基本原理及酶促反应的动力学设计一种分子开关,精确调节目标蛋白表达水平,克服因蛋白过量表达带来的一系列负面效应,维持细胞正常的生理状态,用于研究生物合成途径的代谢节律以及促进生物合成途径高效运转。
细菌的群体感应现象最初是在海洋细菌费氏弧菌Vibrio fischeri中发现的[4]。夏威夷短尾鱿鱼等鱼类的特定器官中特殊共生体V. fischeri的细胞密度很高,达到1010~1011/mL,导致该器官发出荧光;而在海水中游离的V. fischeri,由于密度低于 102/mL,不能发出荧光[5-6]。这种细菌能够感知其数量的变化,当其密度达到一定阈值时才能发生的感应现象称为群感效应[7]。后来的研究发现这种由细菌的细胞与细胞之间的信息交流而引发的群体效应,是由特殊的、微小的、可扩散的化学分子介导的,这些分子被称为自动诱导物或信号分子。细菌正是通过产生、扩散、检测、感应这种化学信号分子从而传递了信息,改变了它们之间的行为,显示出单个细菌所不具有的生理功能和生态特征,包括弧菌调节生物发光、成群浮游形成生物膜现象、抗生素的生物合成等[8-10]。群体感应同时也调控着动植物致病菌的毒性因子的产生,而毒性因子的产生与这些细菌的致病性密切相关[11-12]。
革兰氏阴性细菌V. fischeri的群感效应是以LuxI-LuxR组成的系统进行运作的。LuxR是一种转录调节蛋白,LuxI是信号分子合成酶,其中LuxI能够自动维持低水平地合成可以溶解的小分子信号分子 OHHL (N-3-oxohexanoy1-L-homoserinelactone)[13-14]。当环境中细胞密度较低时,产生的少量信号分子不足以和LuxR结合,因而不能引发依赖LuxR的包含luxI基因在内的荧光操纵子的大量转录。当环境中细胞密度足够大,信号分子浓度达到阈值时,OHHL就进入到细胞内和LuxR结合形成LuxR-OHHL复合物,此复合物结合到PluxI启动子上,使包括luxI基因在内的荧光操纵子得到大量表达,生物体发光。同时,LuxI的增多进一步促进 OHHL的合成,形成一个正反馈调节机制,使荧光功能性基因不断地得到表达,V. fischeri就会发出明显的荧光。此外,科学家也阐明了存在于铜绿假单胞菌和革兰氏阳性细菌的群感效应分子机制[15-19]。
由于群感效应与细胞的致病性密切相关,因此寻找能够抑制细菌群感效应的方法 (即细菌群感猝灭) 对于防病治病具有重要的意义[20-22]。Zhang等首先从芽胞杆菌240B1中分离出了能够降解AHL的酶AiiA,它是由aiiA基因编码的250个氨基酸组成,结构中含有一个保守的HXHXDH锌指结构[23]。2002年 Dong等和 Lee等相继报道了在苏云金芽胞杆菌的不同亚种中广泛存在着aiiA基因,可能原因是苏云金芽胞杆菌在自然生态系统中与革兰氏阴性菌存在竞争关系[24-25]。通过对产物进行化学结构分析发现,该信号分子的内酯环已被打开,由此可知 AiiA是一种AHL内酯酶 (AHL lactonase)[26-27]。
群感效应分子机制的阐明及相关元件的发现为人工设计微生物群落奠定了基础[28-29]。Arnold等利用群感效应设立了一个微生物生物系统,该系统通过调节性自杀效应可以使细胞浓度处于特定的水平[30]。Hasty等设计了一种同步遗传振荡器,利用细菌通过振荡输出信号的方式来构建宏观的生物感应器,为进一步为研究自然界发生的多细胞协调行为的机制提供了一个具体的模型系统[31]。本实验基于细菌群落中普遍存在的群感效应的基本原理并结合酶催化的动力学特征,首先在大肠杆菌群落中建立信号分子高丝氨酸内酯 (AHL) 介导的细胞-细胞交流机制,进而通过控制环境中信号分子 AHL的浓度水平精确控制细胞中靶蛋白的表达水平,实现靶蛋白的定时定量表达,为研究生物合成途径的代谢节律以及促进生物合成途径高效运转提供一种分子工具。
1 材料与方法
1.1 材料
1.1.1 工具酶与试剂
限制性内切酶、ExTaqDNA聚合酶和 T4 DNA连接酶购于宝生物工程 (大连) 有限公司;基因组 DNA提取试剂盒、质粒提取试剂盒、细胞细菌总RNA提取试剂盒、Quant cDNA第一链合成试剂盒购于天根生化科技 (北京) 有限公司;KODTaqDNA聚合酶、SYBR Green Realtime PCR Master Mix购于日本TOYOBO公司;DNA凝胶回收试剂盒购于德国 QIAGEN公司;阿拉伯糖购于北京赛百盛基因技术有限公司;蛋白胨、酵母膏、氯化钠均为国产分析纯试剂;N-(3-oxohexanoyl)-L-homoserine lactone购于TCR试剂公司;海洋肉汤2216购自美国BD公司。
1.1.2 菌株与质粒
苏云金芽胞杆菌Bcillus thuringiensisCGMCC1.792购于中国普通微生物菌种保藏管理中心;V. fischeriES114购于美国菌种保藏中心;pACYC-Duet-1购于Invitrogen公司;E. coliDH5α、E. coliTop10以及质粒 pBAD/Myc-HisA、pET30-Egfp均为本实验室保存。
1.1.3 培养基
BT培养基:蛋白胨10 g,牛肉提取物3 g,NaCl 5 g,蒸馏水定容1 L,pH调为7.0,121 ℃高压蒸汽灭菌20 min,固体培养时加1.5%琼脂。VF培养基:37.4 g海洋肉汤2216溶于蒸馏水,定容至1 L,121 ℃高压蒸汽灭菌20 min。
1.2 方法
1.2.1 培养方法
B. thuringiensis接种到BT培养基中,37 ℃、200 r/min培养16 h。V. fisheri接种到VF培养基中,26 ℃、200 r/min培养16 h。
1.2.2 重组质粒的构建
菌株V. fischeriES114和B. thuringiensisCGMCC1.792的基因组DNA的提取按照细菌基因组DNA提取试剂盒说明书的方法操作。实验中所用的引物见表1,下划线所示为限制性内切酶的酶切位点。
根据NCBI数据库中V. fischeriES114中lux系统基因序列 (GenBank Accession No.AY292966) 设计引物lux for和lux rev。PCR反应条件为:95 ℃预变性5 min;95 1 min℃ ,53 ℃1 min,72 2 min℃ 。PCR产物经BamHⅠ/Hind Ⅲ双酶切后连接到质粒pACYC-Duet-1的对应位点,构建质粒pLux。根据启动子 PluxI、rbs序列和绿色荧光蛋白基因egfp序列设计一组引物,通过叠加延伸 PCR进行 DNA片段拼接,获得 PluxI-rbs-egfp的拼接片段。以 pET30-Egfp为模板,以egfpfor和egfprev为引物进行第一轮扩增;以所得基因片段为模板,以egfpfor2和egfprev为引物进行第2轮扩增;再以所得基因片段为模板,以egfpfor3和egfprev为引物进行第 3轮扩增,最终得到PluxI-rbs-egfp基因片段。该片段经NdeⅠ/Hind Ⅲ双酶切后连接到重组质粒 pLux的对应位点,构建重组质粒pLPE (图1)。该质粒中包含用于靶基因egfp启动表达的全部元件。
以Bcillus thuringiensisCGMCC1.792基因组DNA为模板,简并引物aiiAfor和aiiArev进行℃ ,进行30个循环;72 10 min PCR扩增;PCR反应条件为:95 5 min℃ ;95 ℃1 min,53 1 min ℃ ,进行30个循环;72 10 min℃ ,72 1 min℃ 。PCR产物切胶回收后,用NdeⅠ/XhoⅠ双酶切,连接到经同样酶切处理的表达载体pBAD/Myc-HisA,构建重组质粒pAIIA(图1)。该质粒中包含用于终止靶基因egfp表达的全部元件。

表1 文中所用PCR引物Table 1 PCR primers used in this study

图1 质粒pAIIA和pLPE图谱Fig. 1 Map of plasmid pAIIA and pLPE.
1.2.3 AiiA蛋白质表达以及活性检测
将重组质粒 pAIIA转入到大肠杆菌 Top10中,重组菌接种到LB液体培养基中,加入终浓度为100 μg/mL的氨苄青霉素,37 ℃、200 r/min培养至OD600等于0.6左右时,加入诱导剂L-阿拉伯糖,终浓度为1%,继续诱导培养10 h。离心除去上清液,细胞经洗涤后超声破碎,低温离心后,取上清液用于SDS-PAGE检测蛋白的表达情况。
将1 mLE. coliTop10 (pAIIA) 菌体用生理盐水洗涤 3次后用 495 μL反应缓冲液和 5 μL AHL母液混合,AHL终浓度为2 mmol/L,37 ℃反应10 min,反应液经高速冷冻离心和微孔滤膜过滤处理,HPLC分析色谱柱为Waters C18反向色谱柱,柱温为25 ℃。流动相A液为ddH2O (其中添加终浓度为0.1%乙酸),流动相B液为色谱级乙腈。A液和 B液的比例为 32∶68。高丝氨酸内酯及其产物保留时间分别为 4.9 min和3.9 min。酶的活力定义为每分钟水解 1 µmol AHL所需的菌液。
1.2.4 荧光的镜检
取1 mL过夜培养的重组菌株E. coliTop10(pLPE) 菌液,以E. coliTop10菌液作对照,取2 μL菌液滴在干净的载玻片上,用镊子将盖玻片放在样品上,用吸水纸吸去多余的菌液,放于荧光显微镜下观察,选用100×物镜,10×目镜,先在可见光投射下调出视野,观察细胞形态及密度,拍照后关闭可见光,改换荧光,绿色荧光激发波长为485 nm,拍照保存。
1.2.5 荧光强度的测定
取不同实验条件下的重组菌株E. coliTop10(pLPE/pAIIA) 细胞培养液 200 μL置于96孔板,以E. coliTop10菌液作对照,测定样品在485 nm下的吸光值,相同体积测定3次取平均值。定义相对荧光强度= (重组菌荧光强度-对照菌荧光强度)/对照菌荧光强度。
1.2.6 总RNA的提取及其反转录
不同实验条件下的重组菌株E. coliTop10(pLPE/pAIIA) 的总 RNA提取按细胞细菌总RNA提取试剂盒说明书进行。以经过 DNaseⅠ处理过的 RNA为模板,选取egfpfor1与egfprev1、rrsAfor与rrsArev 2对引物进一步验证总RNA中有无DNA污染。经验证后以无DNA污染的总RNA进行逆转录反应。按照Quant cDNA第一链合成试剂盒说明书进行 RT-PCR,反应体系为:10×RT Mix 2 μL,Super Pure dNTPs(2.5 mmol/L each) 2 μL,Oligo(dT)152 μL,逆转录酶 1 μL,RNase-Free ddH2O 12 μL,模板 RNA 1 μL,总体积 20 μL,反应条件为:37 ℃孵育60 min。
1.2.7 RT-qPCR
以E. coliTop10中rrsA基因为内标[32],检测基因片段大小均为 90 bp左右。按照 SYBR Green Realtime PCR Master Mix试剂盒说明书进行 RT-qPCR。反应体系为:2×SYBR mix 12.5 μL,cDNA 模板 1 μL,上下游引物各 0.25 μL,ddH2O 11 μL,总体积 25 μL。反应条件为:95 ℃ 60 s;95 ℃ 15 s,55 ℃ 15 s,72 ℃ 30 s,循环 40 次。熔解曲线分析20 min,验证是否产生非特异性双链DNA。每个样品重复3次,RT-qPCR结果采用2-△△Ct方法对各个基因的Ct值进行定量分析。
2 结果与分析
2.1 分子开关的设计理念及工作原理
与常规的靶蛋白过表达策略不同,分子开关的设计旨在精确控制细胞内靶基因的蛋白表达水平,实现定量表达的目的,从而有助于研究生物合成途径中关键酶不同的表达水平对该途径代谢节律的影响,也可用于在细胞生长的特定阶段关闭靶基因的表达,从而关闭靶基因所处的代谢通路。为此利用V. fischeriES114中参与群感效应的Lux系统以及信号分子AHL降解酶等生物学元件设计一个可以操控靶基因蛋白表达水平的分子开关。图2显示了构成分子开关的基本元件,工作原理如下:在细胞生长过程中,AHL合成酶编码基因luxI转录,翻译合成蛋白LuxI,后者合成信号分子 AHL。AHL可以自由出入细胞膜,在细胞内和细胞间扩散,当 AHL达到一定浓度时,与 LuxR调控蛋白结合形成复合物LuxR-AHL,该复合物可以上调 PluxI启动子控制下的基因转录,靶蛋白E合成速度加快,此时分子开关处于“on”状态。当需要终止目标蛋白表达时,加入诱导物,启动aiiA转录,翻译合成蛋白质AHL内酯酶,分解AHL,降低环境内AHL的浓度,进而导致LuxR-AHL复合物分离,解除上调效应,此时分子开关处于“off”状态。通过分子开关“on”和“off”状态的切换,可以精确控制靶蛋白的表达水平。为了便于检测分子开关的控制效果,我们选择绿色荧光蛋白EGFP作为靶基因进行验证。此外,考虑到应用的便捷性,我们设计将用于关闭靶基因的元件aiiA和 Lux系统置于同一细胞或不同细胞,并在转录和翻译水平检验分子开关对靶基因表达的实际控制效果。
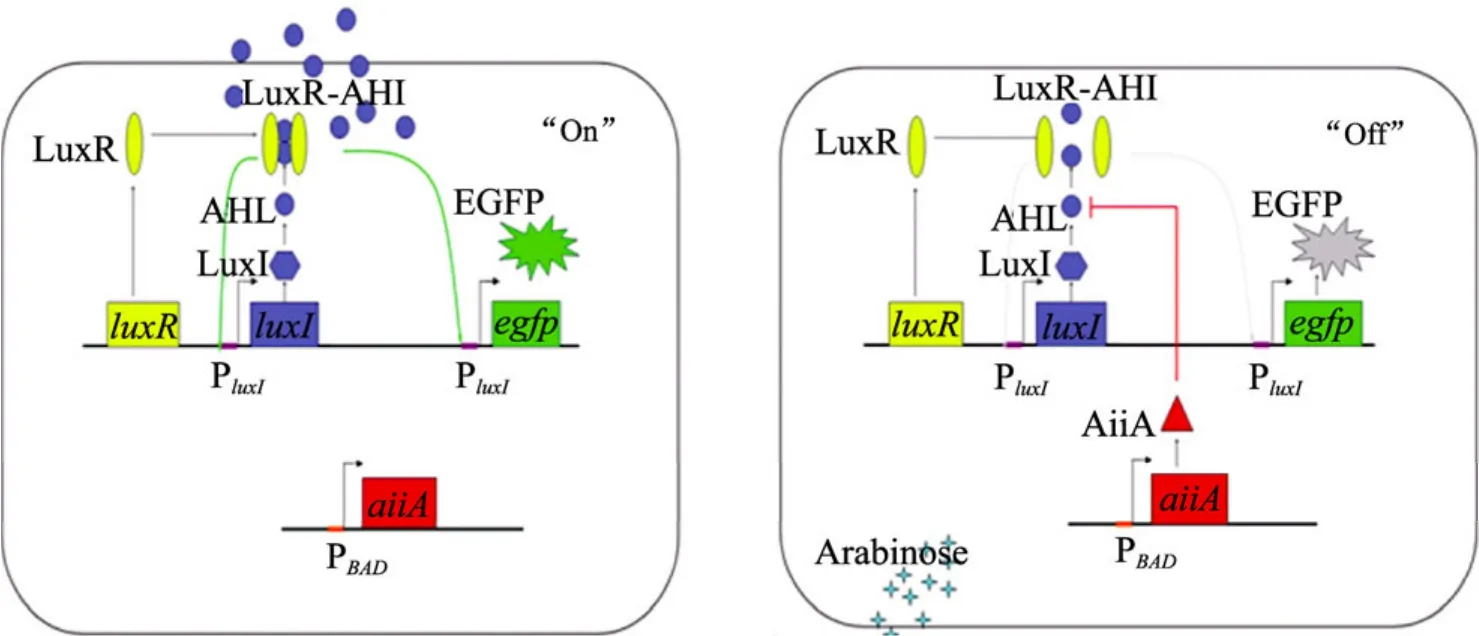
图2 “On”元件、“Off”元件置于同一细胞作用原理示意图Fig. 2 Schematic diagram of molecular switch mechanism.
2.2 分子开关中“Off”元件的构建
2.2.1 aiiA基因的克隆
将文献报道一组功能确认的aiiA基因序列进行同源比对,发现苏云金芽胞杆菌的不同亚种中广泛存在着aiiA基因。根据同源比对结果设计简并引物aiiAfor和aiiArev,以B. thuringiensisCGMCC1.792基因组DNA为模板进行扩增,得到了一条753 bp的基因片段,该片段具有完整的开放阅读框。将翻译的蛋白质序列进行同源比对,发现与芽胞杆菌240B1中分离到的AiiA氨基酸序列的同源性为90%。
2.2.2 aiiA基因在E. coli中表达及酶活性检测
按照方法1.2.1描述成功构建了质粒pAIIA,将质粒转入到E. coliTop10,加入L-阿拉伯糖进行诱导表达,收集全细胞,进行SDS-PAGE凝胶电泳检测。与未诱导细胞相比,诱导后的细胞明显多出一个条带,大小为28 kDa左右,与目的蛋白理论计算值相符,说明aiiA基因可以在E. coli中表达 (图 3)。
按照方法 1.2.2所示的方法检测诱导后E. coliTop10 (pAIIA) 菌体的活性,反应10 min,高速低温离心除去细胞沉淀,上清液经0.22 μm膜过滤后,进行HPLC检测。底物AHL保留时间为 4.9 min,细胞与底物反应生成的产物的保留时间为 3.9 min,检测表明反应液中底物全部耗尽。将产物进行液质分析,确定保留时间为3.9 min的物质为底物AHL内酯环水解后的产物(图4)。上述结果表明诱导生成的AiiA蛋白具有催化活性,全细胞表现出的催化能力为200 μmol/(min·mL) 菌液。

图3 aiiA基因在E. coli中表达的SDS-PAGE图Fig. 3 SDS-PAGE analysis of aiiA gene expression in E. coli. M: protein marker; 1: induced E. coli BL21(pAIIA); 2: uninduced E. coli BL21 (pAIIA).
2.3 分子开关中“On”元件的构建
为了便于检测,本实验选择绿色荧光蛋白EGFP为靶蛋白,将靶基因egfp置于启动子PluxI的控制之下,同时插入核糖体结合序列 rbs。详细的构建过程参见1.2.1。将重组质粒pLPE转入E. coliTop10感受态细胞,挑取单菌落在LB液体培养基中过夜培养,离心肉眼可见菌体呈现亮绿色 (图 5A),通过荧光显微镜可看到细胞发出的绿色荧光 (图5B)。
2.4 蛋白水平检验分子开关对靶基因表达控制

图4 AiiA蛋白催化活力分析Fig. 4 Analysis of AiiA protein catalytic activity. (A)HPLC analysis of AHL. (B) HPLC analysis of reaction product. (C) LC-MS analysis of reaction product.

图5 重组菌株E. coli Top10 (pLPE) 荧光检测Fig. 5 Fluorescence detection of recombinant strain E. coli Top10 (pLPE). (A) Image of the centrifugal cells.(B) Image of the cells observed by fluorescence microscope.
将分子开关“On”和“Off”元件设计在同一细胞中,也即将两种质粒pLPE和pAIIA转入到同一个细胞,构建重组菌E. coliTop10(pLPE/pAIIA)。将分子开关“On”和“Off”元件设计在不同细胞中,也即将两种质粒pLPE和pAIIA分别转入到大肠杆菌 Top10,混合培养E. coliTop10 (pLPE) 和E. coliTop10 (pAIIA)。E. coliTop10 (pLPE/pAIIA) 以及混菌分别接种到含有30 μg/mL氯霉素和100 μg/mL氨苄青霉素的LB液体培养基中,过夜培养后,按0.2%转种量加入到新鲜的100 mL含抗生素的LB培养基中。为了检验分子开关控制EGFP表达效果,分别选择培养初期 (0 h)、对数生长中期 (5 h)加入 1% L-阿拉伯糖诱导物启动 AHL降解酶AiiA的表达,以不添加任何诱导物的培养物为对照。定时取样,测定细胞密度 (OD600),绘制生长曲线。为准确比较细胞内靶蛋白EGFP的蛋白表达水平,将每份样品都稀释到OD600=0.2,用多功能酶标仪测定荧光强度。每个样品测3组平行,计算平均值。图 6A和图 6B显示的分别是重组菌E. coliTop10 (pLPE/pAIIA) 和混菌(E. coliTop10 (pLPE) 和E. coliTop10 (pAIIA))的细胞生长情况以及分子开关对EGFP表达的控制效果。
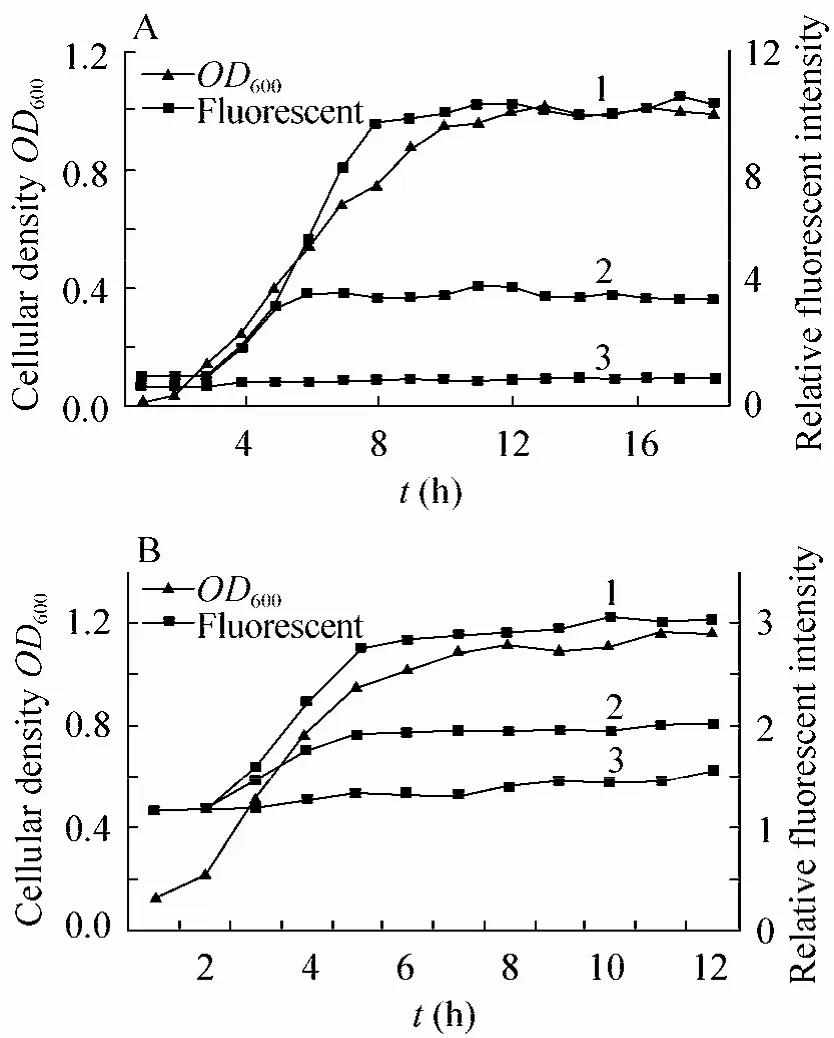
图 6 重组菌株在不同培养条件下的生长曲线及EGFP表达检测Fig. 6 Growth curve and expressed EGFP fluorescence intensity testing of E. coli Top10 (pLPE/pAIIA) (A) and mixture culture of E. coli Top10 (pLPE) and E. coli Top10 (pAIIA) (B) under the different experimental conditions. 1: uninduced; 2: induced at 5 h; 3: induced at 0 h.
从图6A中可见,E. coliTop10 (pLPE/pAIIA)培养过程中,如果不添加诱导剂阿拉伯糖时,分子开关一直处于“On”状态,EGFP的蛋白表达水平与细胞的生产曲线呈正相关性。在细胞处于对数生长期时,EGFP的表达也呈现快速增加,细胞生长进入稳定期后,EGFP的相对荧光强度不再增加,维持在10.5左右。如果在细胞生长初期就加入诱导剂,由于AHL降解酶AiiA的表达导致细胞在生长过程中合成的信号分子 AHL被快速分解,EGFP的表达在整个生长周期中处于非常低的水平,此时分子开关处于“Off”状态。在细胞生长5 h后加入诱导物,可以看到前5 h,EGFP的表达与细胞生产曲线呈正相关性,此后尽管细胞继续生长进入对数生长后期以及稳定期,但EGFP的相对荧光强度基本维持不变。由此可见靶基因的表达的开启与细胞的生长周期密切相关,受群感效应控制,而一旦加入诱导物,可以迅速灵敏地终止靶基因表达。
从图 6B中可见,E.coliTop10 (pLPE) 和E. coliTop10 (pAIIA) 混合培养时,尽管AiiA的表达与靶基因的表达不在处于同一细胞,然而只要启动 AiiA的表达也可以快速关闭靶基因的表达,这与信号分子 AHL可以在细胞间自由扩散直接相关。
由此可见,无论将分子开关中“On”元件、“Off”元件置于同一细胞亦或是不同细胞中,只需通过控制加入诱导剂的时间,可以实现线性定量控制靶蛋白的表达水平。该分子开关具有便捷、灵敏、高效的特点。
2.5 mRNA水平检验分子开关控制效果
以上结果是从蛋白质水平上评估分子开关调控目标蛋白表达的效果,为进一步研究该分子开关的调控效果,通过 RT-qPCR技术检测分子开关中靶基因egfp的转录水平。
E. coliTop10 (pLPE/pAIIA) 在培养过程中,分别在0 h和3 h加入诱导物,以不加诱导物作为对照。在细胞培养过程的不同时间点 (2 h、3 h、3.2 h、3.4 h、3.6 h、4 h 和 10 h) 取样,稀释至相同浓度 (OD600=0.1) 后,提取mRNA,检验无DNA污染后,通过RT-qPCR检查egfp基因的转录水平,选定大肠杆菌的rrsA基因为内参基因。图7显示的是egfp基因分别在不添加诱导物,以及0 h和3 h时添加诱导物后实时检测其mRNA水平的实验结果。由图可见,在细胞培养过程不诱导情况下,egfp基因的相对转录水平在2 h到4 h之间不断提高,此时细胞处于对数生长期,合成代谢较旺盛,基因转录水平较高。10 h时细胞处于生长稳定期,合成代谢降低,基因转录水平也相应降低。细胞生长初期 (0 h) 时加入诱导物,AiiA开始表达,致使AHL信号分子浓度达不到与调控蛋白LuxR结合所需的阈值,所以egfp基因的相对转录水平维持在较低的本底水平。细胞培养3 h后诱导的情况下,egfp基因的相对转录水平在3.2 h比在3 h有所提高,之后逐渐下降。因为刚加入诱导剂,AiiA蛋白的表达需要经过转录和翻译过程,且此时合成的 AiiA蛋白较少,AHL信号分子的合成速度大于降解速度,所以加入诱导剂初期egfp基因的相对转录水平依旧会有所提高。当细胞合成较多的 AiiA蛋白,细胞中信号分子浓度降低,解除上调效应,egfp基因的相对转录水平降低。上述实验结果表明分子开关对靶基因蛋白表达水平的控制是通过间接控制细胞中靶基因mRNA水平实现的。

图7 不同培养条件下egfp基因转录水平的检测Fig. 7 Analysis of the transcriptional level of egfp under the different culture conditions.
3 结论
本实验基于细菌群落中普遍存在的群感效应的基本原理并结合酶催化的动力学特征构建了一种分子开关,并对该分子开关调控目的蛋白表达的效果进行了评估。通过细菌的群感效应启动靶基因表达,通过降解信号分子AHL的群感淬灭效应终止靶基因表达。不论将构成分子开关的“On”元件和“Off”元件设计在同一个细胞内,还是设计在独立的细胞中,分子开关都可以有效地调控目标蛋白的表达。只需通过控制加入诱导剂的时间,就可以实现线性定量控制靶蛋白的表达水平。该分子开关具有便捷、灵敏、高效的特点。本实验设计的分子开关有助于研究生物合成途径中关键酶不同的表达水平对该途径代谢节律,也可用于在细胞生长的特定阶段关闭靶基因的表达,从而关闭靶基因所处的代谢通路。
[1]Liu G, Chater KF, Chandra G, et al. Molecular regulation of antibiotic biosynthesis inStreptomyces. Microbiol Mol Biol Rev, 2013,77(1): 112−143.
[2]Handke P, Lynch SA, Gill RT. Application and engineering of fatty acid biosynthesis inEscherichia colifor advanced fuels and chemicals.Metab Eng, 2011, 13(1): 28−37.
[3]Gosset G.Production of aromatic compounds in bacteria. Curr Opin Biotechnol, 2009, 20(6):651−658.
[4]Cho KW, Colepicolo P, Hastings JW.Autoinduction and aldehyde chain-lendth effects on the bioluminescent emission from the yellow protein associated with luciferase inVibro fischeristrain-Y-1B. Photochem Photobiol, 1989, 50(5):671−677.
[5]Lyell NL, Stabb EV. Symbiotic characterization ofVibrio fischeriES114 mutants that display enhanced luminescence in culture. Appl Environ Microbiol, 2013, 79(7): 2480−2483.
[6]Visick KL, Quirke KP, Mcewen SM. Arabinose induces pellicle formation byVibrio fischeri. Appl Environ Microbiol, 2013, 79(6): 2069−2080.
[7]Fuqua WC, Winans SC, Greenberg EP. Quorum sensing in bacteria: the LuxR-LuxI family of cell density-responsive transcriptional regulators. J Bacteriol, 1994, 176: 269−275.
[8]Nealson KH, Hastings JW. Bacterial bioluminescence: its control and ecological significance. Microbiol Rev, 1979, 43: 496−518.
[9]Eberl L, Winson MK, Sternberg C, et al.Involvement of N-acyl-L-hormoserine lactone autoinducers in controlling the multicellular behaviour ofSerratia liquefaciens. Mol Microbiol,1996, 20: 127−136.
[10]Pierson LS, Keppenne VD, Wood DW. Phenazine antibiotic biosynthesis inPseudomonas aureofaciens30-84 is regulated by PhzR in response to cell density. J Bacteriol, 1994, 176:3966−3974.
[11]Winzer K, Williams P. Quorum sensing and the regulation of virulence gene expression in pathogenic bacteria. Int J Med Microbiol, 2001,291: 131−143.
[12]Majerczyk C, Kinman L, Han T, et al. Virulence ofBurkholderia malleiquorum-sensing mutants.Infect Immun, 2013, 81(5): 1471−1478.
[13]Engebrecht J, Nealson K, Silverman M. Bacterial bioluminescence: isolation and genetic analysis of functions fromVibrio fischeri. Cell, 1983, 32:773−781.
[14]Engebrecht J, Silverman M. Identification of genes and gene products necessary for bacterial bioluminescence. Proc Natl Acad Sci USA, 1984,81: 4154−4158.
[15]Jimenez PN, Koch G, Thompson JA, et al. The multiple signaling systems regulating virulence inPseudomonas aeruginosa. Microbiol Mol Biol Rev,2012, 76(1): 46−65.
[16]Dusane DH, Zinjarde SS, Venugopalan VP, et al.Quorum sensing: implications on rhamnolipid biosurfactant production. Biotechnol Genet Eng Rev, 2010, 27: 159−184.
[17]Thoendel M, Horswill AR. Biosynthesis of peptide signals in gram-positive bacteria. Adv Appl Microbiol, 2010, 71: 91−112
[18]Novick RP, Geisinger E. Quorum sensing inStaphylococci.Annu Rev Genet, 2008, 42: 541−564.
[19]George EA, Muir TW. Molecular mechanisms of agr quorum sensing in virulentStaphylococci.Chembiochem, 2007, 8(8): 847−855.
[20]Tello E, Castellanos L, Duque C. Synthesis of cembranoid analogues and evaluation of their potential as quorum sensing inhibitors. Bioorg Med Chem, 2013, 21(1): 242−256.
[21]Zhu P, Peng H, Ni N, et al. Novel AI-2 quorum sensing inhibitors inVibrio harveyiidentified through structure-based virtual screening. Bioorg Med Chem Lett, 2012, 22(20): 6413−6417.
[22]Galloway WR, Hodgkinson JT, Bowden S,et al.Applications of small molecule activators and inhibitors of quorum sensing in Gram-negative bacteria. Trends Microbiol, 2012, 20(9): 449−458.
[23]Dong YH, Xu JL, Li XZ, et al. AiiA, an enzyme that inactivates the acylhomoserine lactone quorum-sensing signal and attenuates the virulence ofErwinia carotovora. Proc Natl Acad Sci USA,2000, 97: 3526−3531.
[24]Dong YH, Gusti AR, Zhang Q, et al. Identification of quorum-quenching N-acyl homoserine lactonases fromBacillusspecies. Appl Environ Microbiol, 2002, 68(4): 1754−1759.
[25]Sang JL, Park SY, Lee JJ, et al. Genes encoding the N-acyl homoserine thuringiensis lactone-degrading enzyme are widespread in many subspecies ofBacillus thuringiensis. Appl Environ Microbiol,2002,68(8): 3919−3924.
[26]Amara N, Krom BP, Kaufmann GF, et al.Macromolecular inhibition of quorum sensing:enzymes, antibodies, and beyond. Chem Rev, 2011,111(1): 195−208.
[27]Ulrich RL. Quorum quenching: enzymatic disruption of N-acylhomoserine lactone-mediated bacterial communication inBurkholderia thailandensis. Appl Environ Microbiol, 2004,70(10): 6173−6180.
[28]Varga ZG, Szabó MA, Kerényi M, et al.Interference in quorum sensing signal transmission amongst microbial species. Acta Microbiol Immunol Hung, 2012, 59(4): 475−484.
[29]Chen L, Yang L, Duan KM. Bacterial quorum sensing in an evolutionary perspective. Here ditas,2012, 34(1): 33−40 (in Chinese).
陈林, 杨亮, 段康民. 从进化谈细菌细胞间的群体感应信号传递. 遗传, 2012, 34(1): 33−40.
[30]You L, Cox RS, Weiss R, et al. Programmed population control by cell–cell communication and regulated killing. Nature, 2004, 428: 868−871.
[31]Danino T, Palomino OM, Tsimring L, et al. A synchronized quorum of genetic clocks. Nature,2009, 463: 326−330.
[32]Zhou K, Zhou LH, Qing EL, et al. Novel reference genes for quantifying transcriptional responses ofEscherichia colito protein overexpression by quantitative PCR. BMC Mol Biol, 2011, 12: 18.
猜你喜欢
昆明医科大学学报(2021年8期)2021-08-13
江西农业学报(2021年4期)2021-04-20
农村青少年科学探究(2020年5期)2020-08-18
中央民族大学学报(自然科学版)(2018年3期)2018-11-09
新民周刊(2018年8期)2018-03-02
饮食科学(2017年12期)2018-01-02
西南医科大学学报(2015年1期)2015-08-22
少儿科学周刊·少年版(2015年1期)2015-07-07
中国当代医药(2015年9期)2015-03-01
西南军医(2015年6期)2015-01-23
