
集胞藻PCC6803高效基因表达平台的构建与评价
2013-09-04 08:35齐凤霞谈晓明吕雪峰
生物工程学报 2013年9期
齐凤霞,谈晓明,吕雪峰,3
1 中国科学院青岛生物能源与过程研究所 中国科学院生物燃料重点实验室,山东 青岛 266101
2 中国科学院大学,北京 100049
3 山东省能源生物遗传资源重点实验室,山东 青岛 266101
蓝细菌是一种光合原核微生物,可以直接利用太阳能、二氧化碳和水进行生长,并且具有生长速度快、光合作用效率高等优点,因此是一种理想的生物工程宿主,在生物能源、生物制药及生产精细化学品等领域具有广泛的应用前景[1]。
单细胞蓝细菌菌株集胞藻 PCC6803(Synechocystissp. strain PCC 6803) 是研究光合作用机制和蓝藻基因工程的模式生物,可以通过自然转化吸收外源DNA[2],其基因组序列[3]已经于 1996年被测定,是第一个基因组全序列被测定的蓝细菌菌株,遗传背景相对其他蓝细菌较为清晰。近年来,通过基因工程改造集胞藻PCC6803,已经成功生物合成多种高价值分子产品,如天然营养素藻蓝蛋白[4]及食用香料β-石竹烯[5]等,以及化工产品如聚-β-羟丁酸 (PHB)[6]、聚左旋乳酸 (PLA) 前体左旋乳酸[7]、蓝藻胞外多糖 (EPS)[8]、丙酮[9]和异戊二烯[10]等。此外,集胞藻PCC6803也是合成生物燃料的模式基因工程宿主,已经成功合成生物乙醇[11-12]、异丁醇[13]、中长链脂肪酸[14-15]、脂肪醇及脂肪烃[16]等生物燃料产品。
蓝细菌虽然具备可持续生产目的产物的优势,但和传统的生物工程宿主如大肠杆菌相比,蓝细菌对外源基因的表达效率仍然较低,如何使目的基因在蓝藻中得到高效表达是一个关键问题。因此,在蓝细菌中开发和构建高效的基因表达平台,对于蓝细菌代谢工程研究以及提高基因工程蓝细菌的终产物产量具有重要意义。
集胞藻 PCC6803中的外源基因表达载体分为基因组整合载体和穿梭载体,可以通过自然转化[17]或者接合转移[18-19]的方法将目的基因导入蓝细菌细胞内进行表达。基因组整合载体可以将目的基因乃至整个操纵子通过同源整合的方式整合到蓝藻基因组中,其优点是表达稳定,可以克服自主复制质粒不稳定的缺点。而穿梭质粒载体则通过接合转移导入蓝细菌细胞并在细胞质中进行自主复制,其拷贝数常较低,但具有在多个藻株内的穿梭表达的优势。
目前已经有多个启动子在蓝细菌中得到应用,如Susan S. Golden实验室在聚球藻PCC7942外源基因表达平台中使用了光合系统II D1蛋白基因psbA2的启动子PpsbA2[20]以及IPTG诱导性启动子 Ptrc[21]。在集胞藻 PCC6803中得到应用的启动子有 PpsbA2[12,14]、铜离子诱导型启动子PpetE[22-23]以及镍离子诱导性启动子[24]等。同时,Huang等以荧光蛋白基因gfp为报告基因构建并评估了细菌来源的启动子 Ptrc和 Ptac在集胞藻PCC6803中的表达效率[25]。此外,1,5-二磷酸核酮糖缩化酶 (RuBisCO) 是蓝细菌体内主要的可溶性蛋白,因此其启动子 Prbc被认为是一个强启动子并已经成功在聚球藻PCC6301[26-27]、鱼腥藻PCC7120[28]和聚球藻PCC7942[29]中成功应用。然而目前在集胞藻PCC6803中使用rbc启动子表达外源蛋白的报道仅有一例,2011年,Liu等[14]发表在 PNAS的文章中克隆了rbcL基因上游206 bp的片段作为启动子表达硫酯酶Ch FatB2。但是,使用该长度启动子的突变株 (SD232) 并没有成功表达基因并获得目的产物癸酸和辛酸,而使用 PpsbA2启动子表达相同基因的突变株(SD243) 则可以成功合成目的产物,该结果说明集胞藻PCC6803rbcL上游206 bp的片段并不能正常发挥启动子的功能。因此本文尝试在集胞藻PCC6803中重新克隆不同长度的rbc启动子,系统研究其外源基因表达效率,并为基因工程改造蓝细菌筛选有效的启动子元件。
在集胞藻 PCC6803中,RuBisCO的编码基因rbcL、rbcX和rbcS串联在同一个操纵子上,由同一个启动子调控表达 (图1)。Amichay等[30]推断出rbcL上游250 bp的区域为rbc操纵子的启动子元件,并通过测序和序列分析认为在rbcL上游−15~−10区有类似SD序列,并且在翻译起始密码子上游60 bp处发现两处类似−10区和−35区的序列。
在大肠杆菌的研究表明,SD序列以及间隔序列 (SD-AUG) 对基因翻译具有重要影响,改变间隔序列的碱基数目将使得外源蛋白的表达效率发生显著变化[31-32]。SD-AUG的距离同样影响外源蛋白在蓝细菌中的翻译,如 Takeshima等[26]在聚球藻 PCC6301中发现表达产物人类超氧化物歧化酶时,证明 hSOD的比酶活随着SD-AUG间隔变大而显著降低。Deng等[29]在聚球藻PCC7942中表达丙酮酸脱羧酶基因pdc时,保持了rbcLS启动子自身的SD-AUG,将pdc基因与启动子自带的 ATG进行融合表达,结果证明 PDC蛋白表达量提高了近 2倍。而在表达乙醇脱氢酶基因adh时,采用的是adh基因自身的31 bp的SD序列,其蛋白表达量则基本不变。由此证明,使用蓝细菌自身的SD-AUG序列更有利于外源基因在蓝细菌中的表达。
转录终止序列可以终止转录及提高基因的表达效率,在蓝藻中被广泛应用,如 Takeshima等[26]在聚球藻 PCC6301中表达人类超氧化物歧化酶基因hSOD时使用rbc启动子,同时在hSOD基因下游加入rbc的终止序列。Deng等[29]在聚球藻 PCC7942中串联表达pdc和adh两个基因时则在表达元件下游加入了目的基因adh自带的终止序列。
构建有效的遗传改造工具是基因工程蓝细菌生物合成生物燃料等产品的基础。通过选取强启动子和转录终止序列作为有效的基因表达元件,并采用融合表达策略,本文成功构建了可以在集胞藻 PCC6803中高效表达外源基因的一组基因组整合表达平台,并同时构建了可以在多株蓝细菌中表达的穿梭表达平台,并且通过lacZ报告基因测定了表达平台pFQ20的表达效率,以及将该表达平台应用于表达大肠杆菌硫酯酶基因tesA’。
1 材料与方法
1.1 试剂
邻硝基苯 β-D-半乳吡喃糖苷 (ONPG) 购自Sigma-Aldrich公司 (美国)。其他化学试剂购自Merck 公司 (德国) 或 Amresco 公司 (美国)。TaqDNA聚合酶和限制性内切酶购自Fermentas公司(加拿大) 或者TaKaRa公司 (日本)。用于分子克隆的试剂盒购自 Omega公司 (美国) 或 TaKaRa公司 (日本)。DNA引物在Sangon公司 (中国上海) 合成。DNA分子标记购自TaKaRa (日本)。
1.2 集胞藻PCC6803的培养
菌株在 30 ℃条件下进行连续光照培养(30 μE/(m2·s)),突变株 Syn-FQ20 和对照菌株Syn-HB1567在含有10 mg/L壮观素的BG11中培养。用于测定β-半乳糖苷酶活性的藻株在装样量为20 mL的50 mL三角瓶中进行摇床培养,摇动速度为 140 r/min。用于 SDS-PAGE及 Western blotting的藻株,在装样量为300 mL的500 mL三角瓶中进行通气培养。
1.3 质粒和集胞藻突变株的构建
所有引物见表1。以集胞藻PCC6803基因组DNA为模板,以P1/P2和P3/P4为引物对,PCR得到长度分别为0.3 kb和0.2 kb的片段,将这两个片段插入克隆载体pMD18-T (TaKaRa,日本),分别得到质粒pFQ1和pFQ2。
以XbaⅠ和SalⅠ酶切质粒质粒pFQ1,回收0.3 kb片段;将同样以XbaⅠ和SalⅠ酶切pQL4,用 T4连接酶将小片段插入到 pQL4壮观抗性基因Ω[17,19]下游,经过筛选鉴定,得到质粒pFQ5。同样方法,以SmaⅠ-SacⅠ酶切 pFQ2,胶回收0.2 kb的片段插入到酶切pFQ5中,经筛选得到了质粒pFQ6。
用Hind Ⅲ和SacⅠ酶切pFQ6切胶回收获得Ω-Prbc-Trbc(2.5 kb) 片段,利用T4聚合酶补平后插入质粒pKW1188SL[17]的EcoRⅠ位点,后者系用EcoRⅠ切开并补平处理;利用特异引物PCR鉴定目的片段插入方向,正向克隆命名为pFQ9F,反向插入的克隆被命名为pFQ9R (图2)。同理,将补平后的 Ω-Prbc-Trbc插入穿梭质粒pRL59EH[33-34]的HindⅢ位点,筛选鉴定后得到穿梭表达平台 pFQ16 (图1)。
lacZ基因是以大肠杆菌E. coliBL21 (DE3)基因组 DNA为模板,利用引物对lacZ-1/lacZ-2扩增而来,将所得片段插入到 pUC19质粒,得到质粒pQL12;以集胞藻PCC6803基因组DNA为模板,利用P5/P6为引物对进行PCR扩增,得到的1.3 kb的PrbcL片段;用XbaⅠ和SmaⅠ酶切pQL12,胶回收lacZ基因片段,插入到pFQ9F中,得到的质粒再用SalⅠ和XbaⅠ酶切,并用同样酶切PrbcL,利用SalⅠ和XbaⅠ位点,将Prbc替换1.3 kb的PrbcL,得到质粒pFQ20 (图1)。
以大肠杆菌DH5α基因组 DNA为模板,利用QLtesA-1/QLtesA-2为引物,扩增得到0.56 kb的tesA’[35-36],并用NdeⅠ和XhoⅠ酶切后插入到pET21b,得到质粒 pQL5;以pQL5为模板,利用 tesA-1/tesA-3为引物,片段克隆带有 his-tag的片段tesA’-histag并插入到 pMD18-T,得到质粒pFQ4。利用XbaⅠ和SmaⅠ酶切pFQ4,回收tesA’-histag并插入到pFQ20的相同位点,得到表达质粒pFQ25。所有载体构建均经过酶切或者测序验证。将表达质粒按Williams和Zang等[17,37]所述的方法转化集胞藻 PCC6803后,利用含有5 mg/L壮观霉素的BG11琼脂平板筛选转化子,分别得到集胞藻突变株Syn-FQ20和Syn-FQ25。突变株中的外源基因表达元件均通过基因组PCR加以验证。
1.4 集胞藻PCC6803中β-半乳糖苷酶活性测定方法
基于 Miller[38]和 Gao等[22]描述的方法略有改动。取生长对数期 (OD730在1.2~1.5之间) 的培养液进行离心后,将细胞OD730统一调整为1.5。各取1.5 mL上述细胞再次离心收集后,用 1 mL 的 Z Buffer (60 mmol/L Na2HPO4,40 mmol/L NaH2PO4,10 mmol/L KCl,1 mmol/L MgSO4,40 mmol/L β-巯基乙醇) 重悬,依次加入 50 μL的 0.1% SDS、50 μL氯仿反应 1 min使细胞裂解,再加入200 μL ONPG (4 g/L) 并转移至 30 ℃反应 20 min,加入 500 μL的 1 mol/L Na2CO3终止反应。反应液于 12 000 r/min离心2 min将细胞碎片及氯仿沉淀到下层,取1.2 mL上清液测定OD420,以不含藻细胞的反应液作为空白对照。根据公式 Miller=1 000×OD420/(1.5 mL×20 min×OD730) 计算β半乳糖苷酶活性。

表1 本研究中所用的引物Table 1 Primers used in this study
1.5 蛋白印迹法 (Western blotting) 测定外源蛋白的表达
200 mL的集胞藻培养液离心收集,用10 mL 40 mmol/L Tris-Cl (pH 8.0) 重悬藻细胞,加入终浓度1 mmol/L蛋白酶抑制剂PMSF。在冰水浴中超声破碎细胞,每超声30 s间隔30 s~1 min,有效超声时间为5 min。随后,5 000×g离心30 min去除细胞残渣。得到的蛋白样品经过SDS-PAGE和 Western blotting检测。Western blotting采用PVDF膜 (Roche,美国)。利用 6×His-标签探针标记目的蛋白,最终使用碱性磷酸酶显色试剂盒(Amresco,美国) 进行检测。
2 结果与分析
2.1 基因表达元件的克隆与设计
本实验基于不同长度的rbc启动子构建了4个平台质粒pFQ9F、pFQ9R、pFQ16 (图 1) 以及pFQ20 (图2),并选择了其中一个平台用lacZ报告基因比较了与 pHB1567的驱动活性差异。为保证rbc启动子对外源蛋白的翻译效率,本研究采用了一种翻译融合策略,以保持rbc启动子的核糖体结合位点SD序列以及SD序列和ATG的间隔序列 (SD-AUG)。通过PCR克隆了rbc启动子,包涵了rbcL的ATG,并在ATG下游插入一个XbaⅠ位点供外源基因插入,使得目的基因与rbcL的 ATG及酶切位点序列一起融合表达(图1)。在克隆表达外源基因时,需要通过设计引物克隆目的基因片段 (不含起始密码子),将其插入到启动子和终止序列之间。同时选取rbcS终止密码子下游 200 bp的片段作为外源基因表达元件的终止序列Trbc,并在两端设计了酶切位点 (图 1)。

图1 rbc启动子及终止序列克隆示意图Fig. 1 Cloning of rbc promoter and terminator from the Synechocystis rbc operon. Prbc: the 280 bp rbc promoter;PrbcL: the 1 307 bp rbc promoter; Trbc: rbc terminator in Synechocystis; ATG: start codon; XbaⅠ, SalⅠ, SacⅠ, SmaⅠ:the enzyme restriction sites flanked the promoter and terminator fragments. The PCR primers are shown in the figure.Prbc and PrbcL were cloned with the start codon of rbcL and carrying an XbaⅠrestriction site downstream ATG.
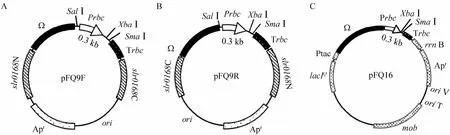
图2 本研究构建的表达质粒图谱Fig. 2 Maps of the constructed gene expression plasmids. (A) pFQ9F: The direction of the expression cassette is forward to slr0168. The target gene fragment (without ATG) can be inserted to the XbaⅠ-SmaⅠsite between the promoter and the terminator. The SalⅠ-XbaⅠsite can be used for promoter replacement. slr0168N: the N terminal of slr0168; slr0168C: the C terminal of slr0168; Apr: ampicillin resistance gene; Ω: Expression cassette for spectinomycin resistance gene; ori: replicon. (B) pFQ9R: the direction of the expression cassette is reverse to that of pFQ9F. (C)pFQ16: the shuttle vector that was designed for conjugative transfer of the expression cassette into other cyanobacteria strains.
2.2 集胞藻PCC6803外源基因表达平台表达β-半乳糖苷酶酶活测定及表达效率对比
本文选择了以pFQ20质粒作为代表,用lacZ报告基因比较了与 pHB1567的驱动活性差异。lacZ作为一种报告基因,具有不影响生物细胞的生长的优点,已经在蓝细菌得到运用[39]。与其他报告蛋白,如荧光素酶[40]和荧光蛋白[41]相比,β-半乳糖苷酶以ONPG为底物进行显色反应,因此可以避免来自蓝细菌细胞内的还原醛以及光合色素等带来的背景干扰,其测定具有显色灵敏和区分度大的优势。因此,本文选用lacZ作为报告基因并检测所构建平台 pFQ20的表达效率(图 3)。实验以含整合有壮观抗性基因的突变株Syn-LY2[42]作为阴性对照,以整合有 PpetE启动子和lacZ报告基因的突变株Syn-HB1567[22]作为阳性对照。在Syn-HB1567中,铜离子诱导型启动子 PpetE在 BG11培养液 (铜离子浓度为316 nmol/L) 驱动表达的 β-半乳糖苷酶活性为71.5 Miller,与已报道结果相近。与之相比,Syn-FQ20菌种中的 β-半乳糖苷酶活性为109 Miller,与对照启动子 PpetE[22]相比,PrbcL启动子表达效率高出52.5%。细胞的生长状态对Miller值有一定的影响,因此本实验中采用对数生长期的细胞 (OD730在1.2~1.5之间) 进行酶活性测定,因此 Miller值比生长中后期 (OD730>1.5) 细胞所测得的更高。
2.3 集胞藻PCC6803表达大肠杆菌硫酯酶
为了检验该表达构建对外源蛋白的表达情况,本文以pFQ20为出发质粒,克隆表达了来自大肠杆菌硫酯酶基因tesA’[35-36],并在蛋白的羧基端加入了一个 6×His-tag标签以追踪本表达平台对外源蛋白的表达情况。

图3 pFQ20质粒结构示意图 (A) 及集胞藻PCC6803突变株中的β-半乳糖苷酶活性(B)Fig. 3 Structure of pFQ20 (A) and the β-galactosidase activity in Syn-FQ20 (B). (A) the lacZ gene was inserted into the pFQ20 using PrbcL as the promoter. (B) Syn-LY2 was used as a negative control, harboring the Ω fragment in slr0168 locus; Syn-HB1567 was used as the positive control applying the copper-induced promoter of PpetE and the lacZ reporter gene. Syn-FQ20 was the mutant strain that transformed by pFQ20.

图4 pFQ25构建 (A)、PCR检测Syn-FQ25突变株的基因型 (B) 以及Western blotting (C) 检测大肠杆菌硫酯酶A在集胞藻PCC6803突变株中的表达Fig. 4 Map of pFQ25 (A) and PCR analysis of the genotype of Syn-FQ25 (B) and Western blotting (C) analysis of E. coli TesA’ expression in Syn-FQ25. (B) M: DNA marker (200 bp DNA Ladder); 1: blank control (PCR without DNA template); 2: negative control (genomic DNA of wild type Synechocystis was used as template for PCR); 3: PCR product using the genomic DNA of Syn-FQ25 as a template, the same primers of slr0168-3 and tesA-1 were used in 1–3; (C) M: protein marker; 2: the negative control of Syn-LY2; 3: positive control using a purified his-tagged enzyme;4–6: triplicate samples of strain Syn-FQ25.
表达质粒 pFQ25 (图 4) 转化集胞藻PCC6803,得到突变株 Syn-FQ25。突变株经粗提蛋白质后经过 SDS-PAGE (浓度为 12%) 和Western blotting检测,结果显示在20 kDa处有显著条带 (图 4)。tesA’-histag长度为 576 bp,其编码蛋白携带 6个组氨酸标签 (6×His-tag) 并在N端加入了两个氨基酸 (XbaⅠ位点编码),总计191个氨基酸,预测分子量为21.7 kDa,所以,最终纯化得到的蛋白与预测的蛋白大小基本相符,说明该表达平台对外源蛋白具有很高的表达效率,可用于在集胞藻 PCC6803中表达外源蛋白。
3 讨论
本文基于不同长度的rbc启动子构建了4个可以用于集胞藻 PCC6803基因表达平台质粒,其中pFQ20表达质粒采用lacZ报告基因比较了与 pHB1567的驱动活性差异,同时利用该质粒表达了外源的tesA’。结果显示,pFQ20对报告基因的表达效率是 109 Miller,同时 Western blotting检测到了 TesA’明显的条带。文献报道rbcL启动子是上游250 bp区域[30],本文构建的rbc启动子 (0.3 kb和1.3 kb) 包含了基因表达必需的元件,可以在蓝细菌中高效稳定表达外源基因的并合成目的产物。例如,Tan等[42]在Metabolic Engineering杂志上发表的蓝细菌合成脂肪醇及脂肪烃的研究采用的就是本文构建的表达平台。其中,pFQ9R被用来表达脂酰还原酶基因far_jojoba,脂肪醇产量为(9.7±2.7) g/(L·OD730),pFQ20被用于串联表达乙酰辅酶 A羧化酶基因accBCDA基因,其脂肪烃产量提高到(161.6±10.3)g/(L·OD730)。以 PpetE为启动子驱动相同基因合成脂肪醇产量为(5.3±1.2) g/(L·OD730),脂肪烃产量提高到(114.9±13.7) g/(L·OD730)[42]。基于本表达平台的突变株中脂肪醇和脂肪烃的产量更高。例如,Gao等[11]利用基于pFQ20表达平台表达了丙酮酸脱羧酶 PDC以及乙醇脱氢酶ADH在集胞藻PCC6803中高效合成乙醇。例如,Gao等利用pFQ20平台成功表达脂酰CoA合成酶 Slr1609[15,43]进行酶学研究并成功测定了其对脂肪族产物的影响。同样,广宿主穿梭质粒pFQ16具有相同的表达元件,又因蓝细菌株系之间利用共用保守的核糖体,因此可以被应用到其他的蓝细菌菌株。本文没有在其他蓝细菌菌株中做进一步效率测定。但已经有报道证明蓝细菌可以利用细菌来源的 Ptrc及 Ptac启动子[35],因此集胞藻 PCC6803来源的启动子在其他蓝细菌中应更有优势。
4 结论
本文通过采用强启动子及强终止序列以及使用翻译融合策略,保护启动子的SD序列及SD与起始密码子之间的间隔序列 (SD-AUG),构建了高效的蓝细菌基因表达平台。本研究构建的表达平台在基因工程改造集胞藻合成生物燃料领域具有重要的应用价值,文中所采用的基因表达策略也为在蓝细菌中高效稳定表达外源基因提供了借鉴。
致谢:感谢中国科学院水生生物研究所徐旭东研究员及高宏副研究员为本研究提供 pHB1567质粒,特致谢意!
[1]Ducat DC, Way JC, Silver PA. Engineering cyanobacteria to generate high-value products.Trends Biotechnol, 2011, 29(2): 95−103.
[2]Grigorieva G, Shestakov S. Transformation in the cyanobacteriumSynechocystissp. 6803. FEMS Microbiol Lett, 1982, 13(4): 367−370.
[3]Nakamura Y, Kaneko T, Hirosawa M, et al.CyanoBase, a WWW database containing the complete nucleotide sequence of the genome ofSynechocystissp. strain PCC 6803. Nucleic Acids Res, 1998, 26(1): 63−67.
[4]Deshmukh DV, Puranik PR. Statistical evaluation of nutritional components impacting phycocyanin production inSynechocystissp. Brazilian J Microbiol, 2012, 43(1): 348−355.
[5]Reinsvold RE, Jinkerson RE, Radakovits R, et al.The production of the sesquiterpene beta-caryophyllene in a transgenic strain of the cyanobacteriumSynechocystis. J Plant Physiol,2011, 168(8): 848−852.
[6]Wu GF, Shen ZY, Wu QY. Modification of carbon partitioning to enhance PHB production inSynechocystissp. PCC6803. Enzyme Microb Technol, 2002, 30(6): 710−715.
[7]Angermayr SA, Paszota M, Hellingwerf KJ.Engineering a cyanobacterial cell factory for the production of lactic acid. Appl Environ Microb,2012, 78(9): 7098−7106.
[8]Ozturk S, Aslim B, Suludere Z. Evaluation of chromium(VI) removal behaviour by two isolates ofSynechocystissp. in terms of exopolysaccha ride(EPS) production and monomer composition.Bioresour Technol, 2009, 100(23): 5588−5593.
[9]Zhou J, Zhang HF, Zhang YP, et al. Designing and creating a modularized synthetic pathway in cyanobacteriumSynechocystisenables production of acetone from carbon dioxide. Metab Eng, 2012,14(4): 394−400.
[10]Lindberg P, Park S, Melis A. Engineering a platform for photosynthetic isoprene production in cyanobacteria, usingSynechocystisas the model organism. Metab Eng, 2010, 12(1): 70−79.
[11]Gao ZX, Zhao H, Li ZM, et al. Photosynthetic production of ethanol from carbon dioxide in genetically engineered cyanobacteria. Energ Environ Sci, 2012, 5(12): 9857−9865.
[12]Dexter J, Fu PC. Metabolic engineering of cyanobacteria for ethanol production. Energ Environ Sci, 2009, 2(8): 857−864.
[13]Varman AM, Xiao Y, Pakrasi HB, et al. Metabolic Engineering ofSynechocystissp. strain PCC 6803 for isobutanol production. Appl Environ Microb,2013, 79(3): 908−914.
[14]Liu, XY, Sheng J, Curtiss III R. Fatty acid production in genetically modified cyanobacteria.Proc Natl Acad Sci USA, 2011, 108(17):6899−6904.
[15]Gao QQ, Wang WH, Zhao H, et al. Effects of fatty acid activation on photosynthetic production of fatty acid-based biofuels inSynechocystissp.PCC6803. Biotechnol Biofuels, 2012, 5(1): 17.
[16]Hu P, Borglin S, Kamennaya NA, et al. Metabolic phenotyping of the cyanobacteriumSynechocystis6803 engineered for production of alkanes and free fatty acids. Appl Energy, 2013, 102: 850−859.
[17]Williams JGK. Construction of specific mutations in photosystem II photosynthetic reaction center by genetic engineering methods inSynechocystis6803.Method Enzymol, 1988, 167: 766−778.
[18]Kreps S, Ferino F, Mosrin C, et al. Conjugative transfer and autonomous replication of a pomiscuous IncQ plasmid in the cyanobacteriumSynechocystisPCC 6803. Mol Gen Genet, 1990,221(1): 129−133.
[19]Fürste JP, Pansegrau W, Frank R, et al. Molecular cloning of the plasmid RP4 primase region in a multi-host-range tacP expression vector. Gene,1986, 48(1): 119−131.
[20]Bustos SA, Golden SS. Expression of thepsbDIIgene inSynechococcussp. strain PCC 7942 requires sequences downstream of the transcription start site. J Bacteriol, 1991, 173(23): 7525−7533.
[21]Mackey SR, Choi JS, Kitayama Y, et al. Proteins found in a CikA interaction assay link the circadian clock, metabolism, and cell division inSynechococcuselongatus. J Bacteriol, 2008,190(10): 3738−3746.
[22]Gao H, Tang Q, Xu XD. Construction of copper-induced gene expression platform inSynechocystissp. PCC 6803. Acta Hydrobiol Sin,2007, 31(2): 240−244 (in Chinese).
高宏, 唐蜻, 徐旭东. 集胞藻 PCC6803铜离子诱导表达平台的构建. 水生生物学报, 2007, 31(2):240–244.
[23]Briggs LM, Pecoraro VL, McIntosh L, et al.Copper-induced expression, cloning, and regulatory studies of the plastocyanin gene from the cyanobacteriumSynechocystissp. PCC 6803. Plant Mol Biol, 1990, 15(4): 633−642.
[24]Liu XY, Curtiss III R. Nickel-inducible lysis system inSynechocystissp. PCC 6803. Proc Natl Acad Sci USA, 2009, 106(51): 21550−21554.
[25]Huang HH, Camsund D, Lindblad P, et al. Design and characterization of molecular tools for a synthetic biology approach towards developing cyanobacterial biotechnology. Nucleic Acids Res,2010, 38(8): 2577−2593.
[26]Takeshima Y, Takatsugu N, Sugiura M, et al.High-level expression of human superoxide dismutase in the cyanobacterium Anacystis nidulans 6301. Proc Natl Acad Sci USA, 1994,91(21): 9685−9689.
[27]Takeshima Y, Sugiura M, Hagiwara H, et al. A novel expression vector for the cyanobacterium,SynechococcusPCC 6301. DNA Res, 1994, 1(4):181−189.
[28]Wang Y, Xu XD. Regulation byhetCof genes required for heterocyst differentiation and cell division inAnabaenasp. strain PCC 7120. J Bacteriol, 2005, 187(24): 8489−8493.
[29]Deng MD, Coleman JR. Ethanol synthesis by genetic engineering in cyanobacteria. Appl Environ Microb, 1999, 65(2): 523−528.
[30]Amichay D, Levitz R, Gurevitz M. Construction of aSynechocystisPCC6803 mutant suitable for the study of variant hexadecameric ribulose bisphosphate carboxylase/oxygenase enzymes.Plant Mol Biol, 1993, 23(3): 465−476.
[31]Gold L, Pribnow D, Schneider T, et al.Translational initiation in prokaryotes. Annu Rev Microbiol, 1981, 35(1): 365−403.
[32]Matteucci M, Heyneker H. Targeted random mutagenesis: the use of ambiguously synthesized oligonucleotides to mutagenize sequences immediately 5' of an ATG initiation codon. Nucleic Acids Res, 1983, 11(10): 3113−3121.
[33]Black TA, Wolk CP. Analysis of a Het-mutation inAnabaenasp. strain PCC 7120 implicates a secondary metabolite in the regulation of heterocyst spacing. J Bacteriol, 1994, 176(8): 2282−2292.
[34]Scholz P, Haring V, Wittmann-Liebold B, et al.Complete nucleotide sequence and gene organization of the broad-host-range plasmid RSF1010. Gene, 1989, 75(2): 271−288.
[35]Davis MS, Solbiati J, Cronan Jr JE. Overproduction of acetyl-CoA carboxylase activity increases the rate of fatty acid biosynthesis inEscherichia coli. J Biol Chem, 2000, 275(37): 28593−28598.
[36]Cho H, Cronan Jr JE. Defective export of a periplasmic enzyme disrupts regulation of fatty acid synthesis. J Biol Chem, 1995. 270(9): 4216−9.
[37]Zang X, Liu B, Liu SM, et al. Optimum conditions for transformation ofSynechocystissp. PCC 6803.J Microbiol, 2007, 45(3): 241.
[38]Miller JH. Experiments in Molecular Genetics.Miller JH, Editor. New York: Cold Spring Harbor Laboratory, 1972.
[39]Schaefer MR, Golden SS. Differential expression of members of a cyanobacterialpsbAgene family in response to light. J Bacteriol, 1989, 171(7):3973−3981.
[40]Kondo T, Strayer CA, Kulkarni RD, et al.Circadian rhythms in prokaryotes: luciferase as a reporter of circadian gene expression in cyanobacteria. Proc Natl Acad Sci USA, 1993,90(12): 5672−5676.
[41]Argueta C, Yuksek K. Summers M. Construction and use of GFP reporter vectors for analysis of cell-type-specific gene expression in Nostoc punctiforme. J Microbiol Methods, 2004, 59(2):181−188.
[42]Tan XM, Yao L, Gao QQ, et al. Photosynthesis driven conversion of carbon dioxide to fatty alcohols and hydrocarbons in cyanobacteria. Metab Eng, 2011, 13(2): 169−176.
[43]Gao QQ, Tan XM, Lu XF, Enzymatic and physiological characterization of fatty acid activation inSynechocystissp. PCC6803. J Basic Microbiol, 2013, doi: 10.1002/jobm.201200228.
猜你喜欢
环球时报(2022-09-20)2022-09-20
舰船科学技术(2022年11期)2022-07-15
江西农业学报(2021年4期)2021-04-20
今日农业(2020年24期)2020-12-15
西藏农业科技(2019年3期)2019-11-04
现代园艺(2018年3期)2018-02-10
上海农业学报(2017年3期)2017-04-10
小资CHIC!ELEGANCE(2015年15期)2015-09-01
西南医科大学学报(2015年1期)2015-08-22
中国当代医药(2015年9期)2015-03-01
