
遗传性疾病PMD的Dys基因多态性探讨
2013-08-20 08:59王银龙沈艳峰王友明魏东敏潘秀兰闫纪琳单铁英耿建芳
中国实用神经疾病杂志 2013年15期
王银龙 沈艳峰 王友明 魏东敏 潘秀兰 闫纪琳 单铁英 耿建芳
河北工程大学附属医院 邯郸 056002
本课题应用聚合酶链式反应-序列特异性引物(PCR-SSP)方法对56例Duchenne型(DMD)及59例Becker型(BMD)两类患者的临床特点及其与Dys基因多态性的相关性进行分析,旨在探讨两类亚型病症的易患因素及防治手段。
1 材料与方法
1.1 临床资料 BMD与DMD患者为1988—2007年我科收集的病例,患者籍贯为长江流域,均为汉族。在有近亲关系的家族中有3例及以上则判定符合诊断标准,在此标准下,BMD患者病例入选59例(17个家系),均为男性。发病年龄13.0~29.0岁,对该组患者临床表征进行数年追踪研究,具体内容有发病年龄、伴发疾病、临床类型及疾病转归和治疗反应性等;DMD患者病例入选56例,均为男性,发病年龄2~5岁无其他自身免疫性疾病或家族史。诊断标准:(1)病态的疲劳程度。(2)肌电图及胸腺CT检查。(3)临床症状的波动性。健康对照组(NC)与2组患者的籍贯相同,均为汉族,无自身免疫性疾病及家族史,共40名。
1.2 实验方法 基因组DNA提取按照提取试剂盒(Promega,美国)的步骤进行。按照参考文献[1]合成Dys1序列特异性引物,通过PCR-SSP方法对Dys进行基因分型,反应体系25μL,利用Toyobo公司Taq酶及缓冲体系,其中DNA含量保持在80~100ng,内参引物2.5μmoL/L,引物3 μmoL/L,Tag酶1U。PCR产物经过1.5%的琼脂糖凝胶电泳,然后再紫外灯下对结果进行拍照。
1.3 统计学分析 组间和组内的发病年龄比较利用t检验,性别和病症亚型构成比采用χ2检验。通过Hardy-Weinberg吻合度测验对估计资料进行可靠性分析。计算各组Dys各等位基因频率,组间基因频率比较采用Fisher检验,当P<0.05,计算相对危险度RR。
2 结果
2.1 遗传性BMD与DMD患者临床资料对比 DMD型患者的临床症状具有较大差异,由于患者依靠轮椅,而造成该病症出现了三种临床亚型[1]。患者在3~5岁就表现出肌无力,于10岁左右需要轮椅,在20岁左右就有高死亡概率。而BMD型患者于13岁左右才会出现肌无力症状,在16岁以后才会借助轮椅,一般寿命在50岁左右,甚至有患者的寿命超过60岁。见表1。
表1 遗传性BMD与DMD患者临床资料对比 (±s,岁)
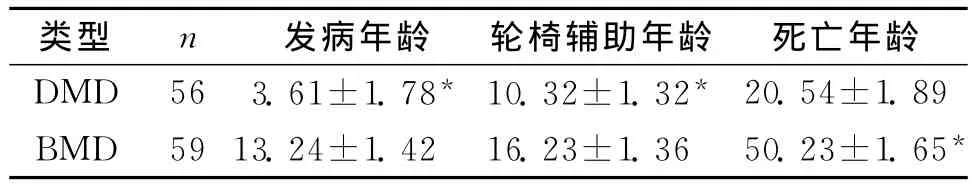
表1 遗传性BMD与DMD患者临床资料对比 (±s,岁)
注:2组比较,*P<0.01
类型 n 发病年龄 轮椅辅助年龄 死亡年龄DMD 56 3.61±1.78* 10.32±1.32*20.54±1.89 BMD 59 13.24±1.42 16.23±1.36 50.23±1.65*
2.2 短串联重复序列(STR)位点的多态性 5个短串联重复序列(STR)的位点均包含CA二核苷酸的重复序列,表2结果显示各位点的片段长度多态性。此表中STR-49共检测到6个多态性片段,长度225~260bp,最具多态性。其次5’DMDⅡ和Intron 45都具有5个多态性片段,长度152~226。Intron 44和Intron 50均具有4个多态性片段,长度178~244。根据此表,通过STR多态性连锁分析法能够作为非缺失型DMD/BMD患者和携带者的诊断方法。见表2。
2.3 BMD/DMD患者外显子缺失突变分析 由表3结果显示,DMD型患者16例在1~36位外显子,36例分布在37~57位外显子,仅4例分布在58~76位外显子。BMD型患者17例在1~36位外显子,39例分布在37~57位外显子,仅3例在58~76位外显子。见表3。

表3 5个STR位点的片段大小多态性
3 讨论
进行性肌营养不良是进行性骨骼肌无力和萎缩为特征的一类遗传性疾病,发病机制具有复杂性,临床表现多样,但共同导致相关肌群的不同程度的肌肉萎缩特征[2]。以肌肉萎缩的发生部位,肌营养不良症可分为6种亚型,分别是Duchenne/Becker型(DMD/BMD)、Emery-Dreifuss型(EMDM)、面-肩-肱型(FSHD)、远端型(Distal Muscular Dystrophy)、肢-带型(LG-MD)、眼咽肌型(OPMD)。而临床上多见于Duchenne/Becker型,其中DMD是临床上发生率最高的X连锁隐性遗传,其患者多在学龄前期出现进行性肌肉无力[3]。据相关统计显示,DMD在新生男婴的发病率为1/3 500,而BMD为1/30 000[4]。近来有研究表明,85%的BMD和65%的DMD患者均有抗肌萎缩基因的缺失。本课题通过分析抗肌萎缩基因缺失外显子的分布位置,初步得出了高缺失概率的外显子位置,为进行缺失突变型BMD/DMD病症的诊断提供了一定的理论基础。
然而,非缺失突变也是进行性肌营养不良的另一原因,约40%的BMD/DMD患者属于抗肌萎缩基因非缺失突变类型,然而针对此类型病症的诊断则成为了当前的一大临床诊断难题[5],而由于STR位点位于dystrophin基因的启动子与内含子中,这些位点具有重复次数多、杂合率较高以及高度多态性的特点,同时这些位点分布于缺失热点区域,所以适合于基因连锁分析,因此通过STR多态性连锁分析法理论上具备了作为诊断非缺失突变类型的可行性,更重要的是,STR多态性连锁诊断能够进行产前基因诊断,进行对BMD/DMD基因携带者的诊断,以预防BMD/DMD遗传疾病。早期诊断对进行性肌营养不良症具有重要的意义,虽然该病症与地域差别无关,但与经济发展水平,社会的文明程度,健康教育水平等状况有密切的关系。一旦大众认识到该病与遗传具有紧密的关系,就会主动与医生合作,加强胎孕检查,预防下一胎携带疾病基因或患病的胎儿出生[6]。另外,还有必要进行扩大范围的筛查本病的基因携带者,如患儿母亲的姐妹,表姐妹及其他有血缘关系的母系女性,这些亲属在生育时,均应该进行相关的遗传病症的筛查检测,以防止相关疾病基因的扩散。
随着分子生物学的深入研究,进行性肌营养不良症从不同类型和亚型的分子机制进行了细致的研究,这一工作的重要性在于为该病症的基因携带者检出,产前诊断,类型区别及明确诊断提供了可行性。伴随人类基因组的草图制图完成,当前已进入基因后时代,进行性肌营养不良的发病机制,该疾病的基因和编码蛋白的分离,以及功能蛋白的研究均将得到进一步的提高。
[1]曹玉红,张光运,张国成,等.Duchenne型进行性肌营养不良40例临床分析 [J].临床床儿科杂志,2008,26(2):96-98.
[2]黄宪章,李艳 .雌激素受体基因多态性的研究进展[J].国外医学临床生物化学与检验学分册,2000,21:254-255.
[3]Parreira SL,Resende MB,Della C,et al.Quantification of muscle strength and motor ability in patients with duchenne muscular dystrophy on steroid therapy[J].Ara Neuropsiquiatr,2007,65(2A):245-250.
[4]Shimomura H,Fujii T,Miyajima T,et al.Prednisolone treatment for Duchenne muscular dystrophy[J].No To Hattatsu,2011,43(1):24-29.
[5]谢琰臣,矫毓娟,许贤豪,等.重症肌无力患者外周血单个核细胞中糖皮质激素受体不同亚型表达和意义[J].中华内科杂志,2007,46:56-59.
[6]Corvol H,Nathan N,Charlier C,et al.Glucocorticoid rcoeptor gene polymorphisms associated with progression of lung disease in young patients with cystic fibrosis[J].Respir Res,2007,8:88-96.
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
皮肤病与性病(2021年3期)2021-07-30
中国生殖健康(2020年4期)2021-01-18
科技资讯(2020年32期)2020-12-28
中国现代神经疾病杂志(2020年9期)2020-01-09
中国心血管杂志(2020年4期)2020-01-09
中国动物检疫(2020年1期)2020-01-08
中国兽药杂志(2019年4期)2019-05-15
中国生殖健康(2018年4期)2018-11-06
