
新型抗菌药利奈唑胺合成的研究
2013-08-14 09:08李桂杰赵静国赵蒙浩
化学与生物工程 2013年1期
李桂杰,赵静国,张 飞,赵蒙浩
(武汉工业学院生物与制药工程学院,湖北 武汉430023)
近年来,随着抗生素的广泛使用甚至滥用,细菌的耐药性日益严重。目前,对耐药性细菌引起的感染性疾病,传统的抗生素药物均无明显的疗效。据世界卫生组织统计,每天约有5万人死于感染性疾病[1]。因此,开发新型抗菌药物成为当务之急。
利奈唑胺Linezolid(化合物Ⅷ,结构式见图1),商品名Zyvox(斯沃),美国辉瑞公司研发,是继磺胺类和喹诺酮类后又一种全新类别唑烷酮类合成抗生素,可用于治疗由需氧的革兰氏阳性菌引起的感染。该类药物结构新颖,作用机制独特,药物作用于细菌蛋白质合成过程中mRNA与50S核糖体亚甲基的起始转译阶段,通过与靠近30S界面的50S亚甲基结合,阻止70S起始复合物的生成[2]。
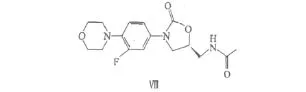
图1 利奈唑胺的结构式Fig.1 The structural formula of linezolid
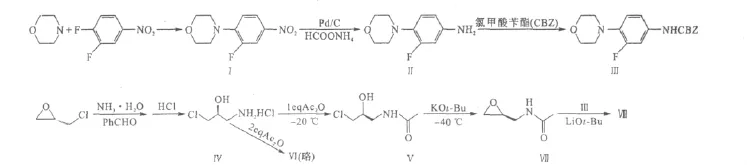
作者以3,4-二氟硝基苯和(S)-环氧氯丙烷为起始原料,根据文献[3],改进了利奈唑胺的合成工艺。合成路线如下:
1 实验
1.1 试剂与仪器
3,4-二氟硝基苯(≥98%),工业级;其余试剂均为国产分析纯。
YRT-3型熔点仪,天津大学精密仪器厂;Mercury Plus-400型核磁共振氢谱仪,美国Varian公司;Trace MS2000型气相质谱联用仪,美国Finnigan公司。
1.2 合成
1.2.1 3-氟-4-吗啉基硝基苯(化合物Ⅰ)
在100mL三口瓶中加入10.4g(120mmol)吗啉、10.1g(100mmol)三乙胺、50mL乙酸乙酯,室温搅拌,缓慢滴加15.9g(100mmol)3,4-二氟硝基苯,反应混合物搅拌过夜,依次用乙酸乙酯萃取、饱和食盐水洗涤、无水硫酸镁干燥,抽滤,旋干溶剂,得淡黄色固体21.6g,收率96.2%,m.p.111~112 ℃(文献值[4]:112~113℃)。
1.2.2 N-苄氧羰基-3-氟-4-吗啉基苯胺(化合物Ⅲ)
在250mL三口瓶中加入11.3g(50mmol)化合物Ⅰ、15.8g(250mmol)甲酸铵、1.1g 10%Pd/C、150 mL丙酮,47℃下搅拌3h,抽滤,滤饼用丙酮洗涤,溶液转移至另一500mL三口瓶中,加入100mL丙酮、7.9mL吡啶,冰水浴,0℃下滴加12.8g(75mmol)氯甲酸苄酯,缓慢升至室温并搅拌过夜,将混合物倾入大量冰水中,抽滤,用少量丙酮洗涤,丙酮与水重结晶,真空干燥,得类白色固体12.9g,收率78.3%,m.p.122~123℃(文献值[4]:122.4℃)。
1.2.3 (2S)-1-氨基-3-氯-2-丙醇盐酸盐(化合物Ⅳ)
在100mL圆底烧瓶中加入30mL乙醇、15.9g(150mmol)苯甲醛、14.7g(420mmol)三乙胺,搅拌1h,冰水浴下滴加13.2g(143mmol)(S)-环氧氯丙烷,缓慢升至室温,过夜,减压浓缩,加入18mL 37%盐酸和20mL水,42℃下反应4h,分出水层,减压浓缩至有固体出现,用甲苯于高真空下反复带水,用无水乙醇和石油醚于-20℃下析晶,抽滤,50℃下真空干燥,得白色晶体16.5g,收率76%,m.p.134~135℃(文献值[5]:125~136℃)。
1.2.4 N-(3-氯-2-羟基)乙酰胺(化合物Ⅴ)
在250mL三口瓶中加入14.6g(100mmol)化合物Ⅳ、50mL甲醇,-30℃下加入11.1g(110mmol)三乙胺,15min后再缓慢滴加25.6g(250mmol)乙酸酐,控制内温不低于-30℃,滴加完毕后缓慢升至室温搅拌3h,减压浓缩,用甲苯和乙醇反复带水,用无水乙醚析出白色固体,抽滤,滤液减压浓缩,得淡黄色油状液体8.91g,收率68.8%。
1.2.5 (R/S)-N-(2,3-环氧丙基)乙酰胺(化合物Ⅶ)
在500mL三口瓶中加入9.1g(60mmol)化合物Ⅴ,用40mL新蒸的四氢呋喃溶解,冷却至-40℃,搅拌下缓慢滴加76mL溶有7.6g(72mmol)叔丁醇钾的四氢呋喃溶液,滴加完毕后,缓慢升至0℃,抽滤,减压浓缩,用四氢呋喃洗涤沉淀,减压浓缩,残留物用柱层析分离(洗脱剂CH2Cl2∶CH3OH=1∶25),得无色粘稠液体5.3g,收率76.2%。
1.2.6 利奈唑胺(化合物Ⅷ)的合成
将0.98g(3mmol)化合物Ⅲ溶于2mL新蒸四氢呋喃中,加入0.19g(6mmol)甲醇,室温、氮气保护下滴加0.48g(6mmol)1mol·L-1LiOt-Bu的四氢呋喃溶液,冰水浴、0 ℃下滴加2.5mL溶有0.41g(3.6 mmol)化合物Ⅶ的四氢呋喃溶液,室温搅拌20h。反应完毕,依次加入2mL饱和氯化铵溶液、15mL水、14mL饱和食盐水溶液、15mL CH2Cl2,分出有机层,水层用CH2Cl2萃取,合并有机层,用无水硫酸镁干燥,蒸干溶剂,用乙酸乙酯-石油醚析晶,得类白色固体0.59g,收率68%。
2 结果与讨论
2.1 利奈唑胺的表征
MS(m/z):337.8[M +];1HNMR(CDCl3,400 MHz),δ:7.43~6.94(m,3H),6.15(t,1H),4.80~4.74(m,1H),4.05(t,1H),3.80(t,4H),3.79(dd,1H),3.72 ~3.55(m,2H),3.02(t,4H),1.99(s,3H)。
2.2 反应条件的选择
文献[5]在合成化合物Ⅰ的过程中需要柱层析分离,本研究发现经下一步还原反应后用丙酮和水重结晶,完全可以除去该步的杂质。
硝基还原过程中,大部分文献采用Pd/C,氢气为还原体系,本研究采用Pd/C,以甲酸铵作供氢剂,通过正交实验确定硝基化合物∶甲酸铵=1∶5(摩尔比)的最佳比例,取得了很好的效果并避免使用氢气带来的安全隐患。
文献[1]采用化合物Ⅲ和化合物Ⅵ合成利奈唑胺,反应过程为在叔丁醇锂的作用下,化合物Ⅵ先脱掉羟基上的乙酰基再脱掉LiCl得到化合物Ⅶ,化合物Ⅶ再与化合物Ⅲ反应得到目标产物利奈唑胺,实验中始终达不到原文献[6]收率,尝试直接将化合物Ⅳ单乙酰化并在-40℃下以叔丁醇钾为催化剂成环得到化合物Ⅶ,虽然比文献多了一步但是收率大幅提高,并且在下一步唑烷酮成环过程中将-78℃滴加丁基锂的苛刻条件改为冰水浴及氮气保护下滴加叔丁醇锂[7,8]。
优化后的工艺路线反应收率较高,操作大大简化,无需苛刻的反应条件,绿色环保。
3 结论
以3,4-二氟硝基苯和(S)-环氧氯丙烷为原料,分别合成两个中间体N-苄氧羰基-3-氟-4-吗啉基苯胺和(R/S)-N-(2,3-环氧丙基)乙酰胺,通过汇聚式合成得到利奈唑胺,总收率约29%。目标产物结构经1HNMR和质谱确证。
[1]Perrault W R,Pearlman A,Godrej D B,et al.The synthesis of N-aryl-5(S)-aminomethyl-2-oxazolidinone antibacterials and derivatives in one step from aryl carbamates[J].Organic Process Research & Development,2003,7(4):533-546.
[3]张慧,杜鲲,桑志培,等.利奈唑胺的合成[J].中国医药工业杂志,2011,42(4):245-247.
[4]Brickner S J,Hutchinson D K,Barbachyn M R,et a1.Synthesis and antibacterial activity of U-100592and U-100766,two oxazolidinone antibacterial agents for the potential treatment of multidrug-resistant gram-positive bacterial infections[J].J Med Chem,1996,39(3):673-679.
[5]赵肖玉,马燕如,徐正.利奈唑胺合成工艺的改进[J].华西药学杂志,2007,22(2):179-181.
[7]Poulakou G,Giamarellou H.Investigational treatments for postoperative surgical site infections[J].Expert Opin Investig Drugs,2007,16(2):137-155.
[8]Jung Young Hoon,Kim Chang Min.A formal asymmetric synthesis of mugineic acid:An efficient synthetic route through chiral oxazolidinone[J].Medicinal Chemistry,1999,22(6):624-628.
猜你喜欢
化工设计(2022年4期)2023-01-02
化工技术与开发(2022年4期)2022-04-22
广州化工(2022年3期)2022-02-24
理化检验-化学分册(2021年4期)2021-04-18
农药科学与管理(2019年8期)2019-11-23
农药科学与管理(2019年12期)2019-05-20
科技创新与品牌(2019年12期)2019-02-06
化工管理(2017年35期)2018-01-10
天津化工(2015年4期)2015-12-26
化学工业与工程(2015年1期)2015-02-10
