
NiS-PdS/CdS光催化剂的水热法合成及其可见光分解水产氢性能
2013-06-23 06:51林培宾高寒阳陈小平上官文峰
物理化学学报 2013年6期
林培宾 杨 俞 陈 威 高寒阳 陈小平 袁 坚 上官文峰
(上海交通大学燃烧与环境技术中心,上海200240)
1 引言
光催化分解水产氢技术能有效地将太阳光直接转化为氢能,越来越受到人们的关注.1-3在几十年的发展中,大量光催化剂得到了研究和应用,但大多数只能响应占太阳能4%左右的紫外光.2,4,5为了提高太阳能转化效率,高效响应可见光的光催化剂的研究十分必要.6
在光解水催化剂中,CdS具备合适的禁带宽度和导带位置,是一种可见光响应型催化剂.7为了克服其光溶性(光腐蚀性),8,9并提高产氢效率,人们通过负载各种贵金属(如Pt,Pd,Rh等)10,11及其氧化物(如RuO2等)12进行修饰和改性.一些非贵金属助剂(如WS213和MoS214等)也被报导,但产氢效率不高.最近,Li课题组15报道了利用共负载双贵金属的Pt-PdS/CdS光催化剂,获得了很高的产氢活性和量子效率.Bao等16通过两步法合成了多孔纳米片和空心纳米棒CdS,负载Pt后也获得了较高的催化活性.
本文作者制备了不含Pt的CdS基双负载型光催化剂NiS-PdS/CdS,结果表明通过水热法合成的这一光催化剂具有较高的产氢活性.本文对该光催化剂的制备方法、结构特性和光解水活性进行了研究,并对其作用机理等作了初步讨论.
2 实验部分
2.1 材料制备
所用的试剂:醋酸镉 (≥98.5%)、硫化钠(≥98.0%)、乳酸(≥85.0%)、无水亚硫酸钠(≥97.0%)、醋酸镍(≥98.0%)、氯化钯(含 Pd 量≥59.0%)、硫脲(≥99.0%)均为分析纯,由国药集团化学试剂有限公司生产.所有试剂未作进一步纯化.
2.1.1 CdS的制备
取Na2S溶液缓慢地加入Cd(OAc)2溶液中,两者物质的量比为4:5.溶液剧烈搅拌24 h.将以上溶液静置24 h,然后清洗过滤,得到沉淀物CdS,将其转入180 mL高压反应釜,在200°C下水热24 h,最后用水和乙醇多次清洗过滤,80°C真空干燥24 h制得产物,标记为CdS(HT-24h).CdS(HT-72h)的制备方法与上述方法相同,水热时间为72 h.
2.1.2 NiS-PdS/CdS的制备
在80 mL高压反应釜中依次加入0.3 g CdS(HT-24h)、1 g硫脲、5 mmol·L-1醋酸镍、氯化钯(Pd含量为0.00236 g·mL-1)和50 mL纯水.剧烈搅拌1 h后140°C水热5 h,最后用水和乙醇多次清洗过滤,80°C真空干燥6 h制得NiS-PdS/CdS(HT-24h).以相同方法制备得到了NiS/CdS(HT-24h)、PdS/CdS(HT-24h)和NiS-PdS/CdS(HT-72h).
2.2 催化剂表征
X射线粉末衍射(XRD)分析在日本理学RigakuX射线粉末衍射仪(CuKα靶,λ=0.15406 nm,40 kV,20 mA)进行.紫外-可见漫反射光谱(DRS)分析在日本岛津公司UV-2450紫外-可见分光光度计上进行.透射电镜(TEM/HRTEM)分析在日本电子株式会社/英国OXFORD公司的JEM-2010/INCA OXFORD分析型透射电子显微镜上进行,加速电压为200 kV.荧光(PL)瞬态测量在美国铂金埃尔默仪器有限公司的LS-50B型荧光分光光度计上进行.
2.3 光催化剂的活性评价
光解水产氢性能测试在自制的真空俯照式PYREX玻璃反应器(350 mL)中进行.反应前将0.15 g光催化剂样品及80 mL乳酸牺牲剂(30%(体积分数))17,18加入到反应器中,然后将反应器抽成真空.反应中利用磁力搅拌使光催化剂更好地分散,并用冷却水维持反应体系处于室温(20-25°C).光源采用北京中教科技有限公司300 W氙灯,利用滤光片(λ>420 nm)将紫外光滤去.生成的气体由气相色谱定量检出(华爱色谱9160,TCD检测器,5A分子筛填充柱,氩气为载气,柱温40°C).
光催化剂的量子效率根据国家标准(GB/T 26915-2011)进行测试.选取0.2 g样品分散于200 mL乳酸牺牲剂(30%)中.氙灯光源电流为18 mA.采用外置式光催化制氢反应器进行产氢.产氢反应前通N215 min.利用420 nm带通滤光片照射样品前,预先使用全波段照射1 h.测量过程中,定时用100µL微型进样器取样注入气相色谱,检测样品气中氢气的含量,得到产氢数据从而计算参与反应的电子数.采用光纤光谱仪(型号为AvaSpec-2048-usbz)测试对应光斑光强,然后计算入射的光子数.最后根据公式(1)计算表观量子效率(φH2):

其中,阿伏加德罗常数N0=6.02×1023;RH2为单位时间的氢产量(mol·s-1);n为单位时间的入射光子数.
3 结果与讨论
3.1 XRD结果分析
图1为催化剂CdS、CdS(HT-24h),NiS-PdS/CdS(HT-24h)和NiS-PdS/CdS(HT-72h)的XRD光谱图.可以看出未经水热处理的CdS为立方相.经过水热处理后,部分立方相的CdS转化为六方相.NiS-PdS/CdS(HT-72h)已完全转化为六方相,其中2θ为24.6°、26.6°、28.3°的特征峰分别对应六方相CdS的(100)、(002)、(101)晶 面.CdS(HT-24h)和 NiS-PdS/CdS(HT-24h)的XRD图说明两者为立方相和立方相的混合物.共负载NiS和PdS后,NiS-PdS/CdS(HT-24h)的衍射峰锐化,表明CdS(HT-24h)的结晶度提高.XRD并未检测到NiS和PdS的衍射峰,这可能是NiS和PdS含量较少且在CdS(HT-24h)表面上的高度分散所致.
3.2 紫外-可见漫反射分析
图2为催化剂CdS,CdS(HT-24h)和NiS-PdS/CdS(HT-24h)的吸收光谱图.可以看出,CdS(HT-24h)吸收边约540 nm.共负载NiS和PdS后,NiS-PdS/CdS(HT-24h)吸收边未有明显改变,但在更宽的可见光区域呈现出较大的吸收,这主要是表面负载物NiS和PdS对可见光吸收的作用所致.
3.3 TEM分析
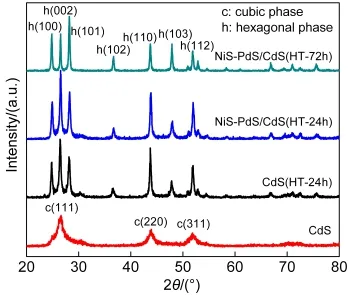
图1 CdS和NiS-PdS/CdS的XRD图Fig.1 XRD patterns of CdS and NiS-PdS/CdS

图2 CdS,CdS(HT-24h)和 NiS-PdS/CdS(HT-24h)的紫外-可见漫反射光谱(DRS)Fig.2 UV-Vis diffuse reflectance spectra(DRS)of CdS,CdS(HT-24h),and NiS-PdS/CdS(HT-24h)
图3为CdS(HT-24h)和NiS-PdS/CdS(HT-24h)的TEM和HRTEM图.从图3(a)可看出,CdS(HT-24h)主体形貌是球状结构,颗粒大小均匀,粒径在30-40 nm之间.复合负载后的NiS-PdS/CdS(HT-24h)样品(图3(b))与CdS(HT-24h)相差不大,说明进一步水热并没有增加CdS(HT-24h)的粒径.图3(c)是NiS-PdS/CdS(HT-24h)透射电镜的放大照片,图中晶格间距为0.337、0.321和0.296 nm,分别对应CdS的(022)面、PdS的(200)面和NiS的(100)面,证实了Pd和Ni是以PdS和NiS的形式负载在CdS的表面上.
3.4 瞬态荧光分析
图4 为CdS(HT-24h)、NiS/CdS(HT-24h)、PdS/CdS(HT-24h)和NiS-PdS/CdS(HT-24h)的瞬态荧光图谱及拟和曲线图,表1为拟合衰减曲线后的平均寿命计算结果.瞬态荧光技术能够通过计算荧光寿命反映载流子的迁移速率,荧光寿命越短,其迁移率也就越快,载流子的分离也就越好.19,20为了对比各样品的电子空穴分离效率,分别取0.003 g的样品与0.057 g的KBr混合、碾磨,然后在7 MPa压3 min,得到厚度相等的均匀薄片.通过荧光测试、衰减图谱拟合以及计算可知,NiS-PdS/CdS(HT-24h)、NiS/CdS(HT-24h)、PdS/CdS(HT-24h)CdS(HT-24h)的荧光寿命分别为1.30、1.42、1.49、1.95 ns.NiS-PdS/CdS(HT-24h)具有最短的荧光寿命,意味着共负载相对于单独负载NiS或PdS,光生电子和空穴更容易迁移和分离,因此NiS-PdS/CdS(HT-24h)可能具有最高的活性.

图3 (a)CdS(HT-24h)的TEM图;NiS-PdS/CdS(HT-24h)的(b)TEM图和(c)HRTEM图Fig.3 (a)TEM image of CdS(HT-24h),(b)TEM and(c)HRTEM images of NiS-PdS/CdS(HT-24h)
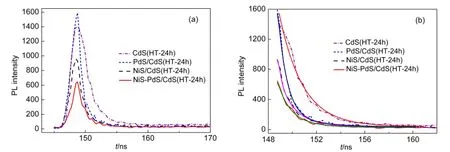
图4 CdS(HT-24h),NiS/CdS(HT-24h),PdS/CdS(HT-24h)和NiS-PdS/CdS(HT-24h)的(a)瞬态荧光(TRPL)光谱及(b)拟和曲线Fig.4 (a)Transient photoluminescence(TRPL)spectra and(b)fitting curves of CdS(HT-24h),NiS/CdS(HT-24h),PdS/CdS(HT-24h),and NiS-PdS/CdS(HT-24h)

表1 根据图4数据计算的各样品的平均荧光寿命Table 1 Averaged PLlifetime of samples calculated from Fig.4
3.5 光催化制氢活性
图5 对 比 了 CdS(HT-24h)、NiS/CdS(HT-24h)、PdS/CdS(HT-24h)、NiS-PdS/CdS(HT-24h)和 NiS-PdS/CdS(HT-72h)五种催化剂的产氢反应活性.可以看到:样品的产氢活性顺序为NiS-PdS/CdS(HT-24h)>NiS-PdS/CdS(HT-72h)>PdS/CdS(HT-24h)>NiS/CdS(HT-24h)>CdS(HT-24h).单独负载NiS或PdS都能提高CdS(HT-24h)的催化活性,但提升不大,但是当同时负载NiS和PdS时,CdS(HT-24h)催化活性显著提高,当NiS和PdS负载量分别为1.5%和0.41%(w)时,活性达到6556 μmol·h-1,催化活性是未负载时的7倍,是NiS/CdS(HT-24h)的近3倍.测得表观量子效率达到47.5%(λ=420 nm).共负载后活性的提升可能是因为NiS和PdS在CdS(HT-24h)表面能起到协同促进作用,使得光生电子空穴对能更有效地分离并转移到催化剂表面.另外,NiS-PdS/CdS(HT-24h)的活性明显好于NiS-PdS/CdS(HT-72h).这可能是CdS不同的相结构造成的.混合相的CdS由于各相存在光电性能差异,形成的异质结结构可以促进电子-空穴对的有效分离,21因此可以提高产氢活性.
3.6 NiS和PdS负载量对产氢活性的影响
在CdS光解水制氢实验中,选用CdS(HT-24h)作为光催化剂,通过控制加入不同量的氯化钯溶液和醋酸镍溶液来比较两种助催化剂负载量对CdS光催化活性的影响.

图5 样品的可见光产氢活性Fig.5 Hydrogen production activity of samples under visible light irradiation
图6(a)为不同PdS负载量的CdS(HT-24h)每小时平均产氢量.可以看出,当负载PdS后,CdS的活性明显提高.仅仅负载0.1%(w)的PdS时,CdS的活性增长了近3倍.随着PdS负载量增大,CdS催化活性逐步提高.当负载0.41%(w)PdS时,CdS具有最好的催化活性.当PdS负载量继续增加时,产氢量呈下降趋势,这可能是因为负载PdS过多会影响CdS表面性质和光吸收能力.
在确定0.41%(w)PdS最佳值的基础上,图6(b)给出了不同NiS负载量的NiS-PdS/CdS的产氢速率.NiS-PdS/CdS负载NiS后,产氢量增加明显,而且随着NiS负载量增大而提高.当NiS负载量为1.5%(w)左右时,NiS-PdS/CdS表现出最高的产氢速率.继续提高负载量,活性平缓下降,但仍然维持较高的活性.当负载量超过2.5%(w)后,NiS-PdS/CdS活性有明显的下降.这可能是因为负载过多NiS会减少CdS表面的活性位数量,并且会起遮光作用,影响CdS对光的吸收,从而导致活性下降.
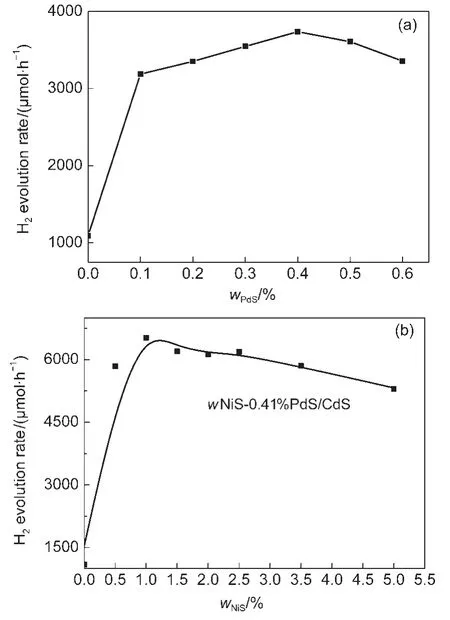
图6 NiS-PdS/CdS(HT-24h)催化活性与(a)PdS及(b)NiS负载量的关系Fig.6 Relationship between the photocatalytic activity and the loading amount of(a)PdS or(b)NiS
NiS-PdS/CdS具有高效催化产氢效率来自于共负载的NiS和PdS,并且两者起着不同的作用.NiS的导带位置(0.53 eV(vsNHE(normal hydrogen electrode)))远低于H+/H2的还原电位(0 eV(vsNHE)),因此NiS本身无法通过光生电子来光解水产氢,但已有研究报道,NiS负载在催化剂表面时,具有较低的势垒,有利于还原水产氢反应的进行,4,17,22而且在促进光生电子转移和H2电化学解吸附方面发挥出重要作用.23NiS催化还原水中的H+的过程可表示为公式(2)和(3).17
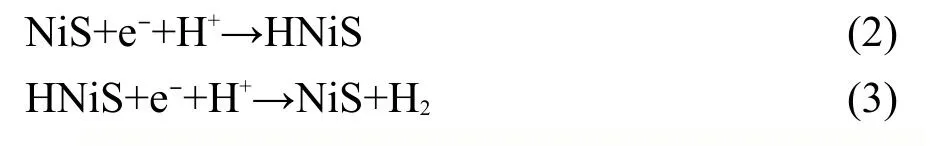
PdS的价带位置为1.34 eV(vsNHE)左右,稍高于CdS的价带位置(+1.50 eV),因此CdS内的光生空穴能容易地转移到PdS价带上,使空穴与电子复合的几率降低,从而提高催化活性和稳定性.15,24
为了进一步探讨NiS、PdS在光催化中的作用,我们采用NiS和PdS分别与Pt进行复合,利用对比实验进行考察.
图7比较了Pt/CdS,NiS/CdS,PdS/CdS,Pt-NiS/CdS,Pt-PdS/CdS及NiS-PdS/CdS催化剂的催化活性.由图可以看出,NiS-PdS/CdS的产氢速率最大.
Pt助催化剂因为具有最高的功函数,从而被公认为是有效的析氢助剂(还原位).25当Pt与NiS复合后,其活性与Pt或NiS单独负载相比,并没有明显提高;而当Pt与PdS复合后,活性大大高于二者的单独负载.这说明NiS、PdS在光催化过程中的作用是不同的:NiS与Pt类似,具有传递电子的作用,当与Pt复合后,二者存在竞争作用,而PdS具有传递空穴的作用,与Pt复合后,电子和空穴分别在Pt和PdS富集,从而更有利于载流子的分离,提高活性.当NiS与PdS负载于CdS后,NiS与PdS分别作为还原位和氧化位,极大提高了产氢活性.值得注意的是,本研究中NiS-PdS/CdS的催化活性要好于Pt-PdS/CdS,这可能与水热法制备后NiS和CdS之间良好的接触面以及NiS的表面特性有关.相对于单独负载,复合后的高活性说明通过构建二元甚至多元助催化剂同时传递电子和空穴是进一步提高光催化活性的有效手段.

图7 负载不同助催化剂的CdS(HT-24h)的光催化活性Fig.7 Photocatalytic activity of CdS(HT-24h)loaded with various cocatalysts
4 结论
本研究利用水热法制得了NiS-PdS/CdS共负载型复合光催化剂.NiS和PdS很好地分散在CdS表面.共负载能提高CdS的光生载流子的迁移和分离能力,当NiS和PdS浓度分别在1.5%和0.41%(w)时,NiS-PdS/CdS活性最好.0.15 g NiS-PdS/CdS(HT-24h)在乳酸牺牲剂中的产氢量为6556 μmol·h-1,是未负载CdS活性的7倍,是NiS/CdS的近3倍,表观量子效率为47.5%(λ=420 nm).NiS和PdS在光催化剂中分别起还原助剂和氧化助剂的作用,因而明显增强光解水产氢活性.
(1) Osterloh,F.E.Chem.Mater.2008,20,35.doi:10.1021/cm7024203
(2) Kudo,A.;Miseki,Y.Chem.Soc.Rev.2009,38,253.doi:10.1039/b800489g
(3)Maeda,K.;Domen,K.J.Phys.Chem.C2007,111,7851 doi:10.1021/jp070911w
(4)Kato,H.;Asakura,K.;Kudo,A.J.Am.Chem.Soc.2003,125,3082.doi:10.1021/ja027751g
(5) Chen,W.;Gao,H.Y.;Yang,Y.;Lin,P.B.;Yuan,J.;Shangguan,W.F.;Su,J.C.;Sun,Y.Z.Acta Phys.-Chim.Sin.2012,28,2911.[陈 威,高寒阳,杨 俞,林培宾,袁 坚,上官文峰,苏佳纯,孙洋洲.物理化学学报,2012,28,2911.]doi:10.3866/PKU.WHXB201208011
(6) Murphy,A.B.;Barnes,P.R.F.;Randeniya,L.K.;Plumb,I.C.;Grey,I.E.;Horne,M.D.;Glasscock,J.A.Int.J.Hydrog.Energy2006,31,1999.doi:10.1016/j.ijhydene.2006.01.014
(7) Chen,X.P.;Shangguan,W.F.Front.Energydoi:10.1007/s11708-012-0228-4
(8)Wen,F.Y.;Yang,J.H.;Zong,X.;Ma,Y.;Xu,Q.;Ma,B.J.;Li,C.Progress in Chemistry2009,21,2285.[温福宇,杨金辉,宗 旭,马 艺,徐 倩,马保军,李 灿.化学进展,2009,21,2285.]
(9) Williams,R.J.Chem.Phys.1960,32,1505.doi:10.1063/1.1730950
(10)Sathish,M.;Viswanathan,B.;Viswanath,R.P.J.Hydrog.Energy2006,31,891.doi:10.1016/j.ijhydene.2005.08.002
(11) Li,Y.X.;Xie,Y.Z.;Peng,S.Q.;Lu,G.X.;Li,S.B.Chemosphere2006,63,1312.doi:10.1016/j.chemosphere.2005.09.004
(12) Sakata,T.;Hashimoto,K.;Kawai,T.J.Phys.Chem.1984,88,5214
(13)Zong,X.;Han,J.F.;Ma,G.J.;Yan,H.J.;Wu,G.P.;Li,C.J.Phys.Chem.C2011,115,12202.doi:10.1021/jp2006777
(14)Zong,X.;Yan,H.J.;Wu,G.P.;Ma,G.J.;Wen,F.Y.;Wang,L.;Li,C.J.Am.Chem.Soc.2008,130,7176.doi:10.1021/ja8007825
(15)Yan,H.J.;Yang,J.H.;Ma,G.J.;Wu,G.P.;Zong,X.;Lei,Z.B.;Shi,J.Y.;Li,C.J.Catal.2009,266,165.doi:10.1016/j.jcat.2009.06.024
(16)Bao,N.Z.;Shen,L.M.;Takata,T.;Domen,K.Chem.Mater.2008,20,110.doi:10.1021/cm7029344
(17)Zhang,W.;Wang,Y.;Wang,Z.;Zhong,Z.;Xu,R.Chem.Commun.2010,46,7631.doi:10.1039/c0cc01562h
(18) Harada,H.;Sakata,T.;Ueda,T.J.Am.Chem.Soc.1985,107,1773.doi:10.1021/ja00292a060
(19) Lin,K.;Chuang,C.;Lee,Y.;Li,F.;Chang,Y.J.Phys.Chem.C2012,116,1550.doi:10.1021/jp209353j
(20) Spanhel,L.;Weller,H.;Henglein,A.J.Am.Chem.Soc.1987,109,6632.doi:10.1021/ja00256a012
(21)Hurum,D.C.;Agrios,A.G.;Gray,K.A.J.Phys.Chem.B2003,107,4545.doi:10.1021/jp0273934
(22) Zou,Z.G.;Ye,J.H.;Sayama,K.;Arakawa,H.Nature2001,414,625.doi:10.1038/414625a
(23)Assuncao,N.;Giz,M.;Tremiliosi,G.;Gonzalez,E.J.Electrochem.Soc.1997,144,2794.doi:10.1149/1.1837897
(24)Yang,J.H.;Yan,H.J.;Wang,X.L.;Wen,F.Y.;Wang,Z.J.;Fan,D.Y.;Shi,J.Y.;Li,C.J.Catal.2012,290,151.
(25) Min,S.X.;Lü,G.X.Acta Phys.-Chim.Sin.2011,27,2178.[敏世雄,吕功煊.物理化学学报,2011,27,2178.]doi:10.3866/PKU.WHXB20110904
猜你喜欢
化学工程师(2023年1期)2023-02-17
科学之友(2022年11期)2022-11-03
工业水处理(2022年6期)2022-06-23
石油化工高等学校学报(2021年3期)2021-07-15
理化检验-化学分册(2020年12期)2021-01-26
上海农业科技(2019年1期)2019-02-22
中国果业信息(2018年5期)2018-01-17
无机盐工业(2017年5期)2017-05-25
郑州大学学报(理学版)(2017年1期)2017-04-07
物理化学学报(2017年3期)2017-03-11
