
质谱电喷雾电离源研究新进展
2013-05-29 05:54李佳斌郝斐然张养军
质谱学报 2013年2期
李佳斌,郝斐然,田 芳,张养军
(蛋白质组学国家重点实验室,北京蛋白质组研究中心,军事医学科学院放射与辐射医学研究所,北京 102206)
质谱是一种广泛使用的测量带电粒子质荷比的分析技术,通过测定质荷比可以获得粒子的质量,进而鉴定被分析物质的分子组成,揭示诸如肽段等化合物的化学组成和结构,还可通过质谱峰面积的测量实现被分析物质的定量,其特点是高灵敏度和高选择性。质谱仪器主要由离子源、质量分析器、检测器和真空系统构成,其中最为关键的部件是离子源,其作用是使待分析物离子化。不同的离子源应对的分析物不同,如ESI源和MALDI源一般适用于极性物质的分析,并且主要针对液体样品。最近Zhang等[1]提出的LTP(低温等离子探针)源既可直接用于气态样品离子化,又可用于固态样本解析电离。由于ESI源和MALDI源的软电离特性,使得对非挥发性、受热易分解物质的分析成为可能,受到生物大分子和药物研究领域科学家的关注,并因其对生命科学发展的巨大贡献,ESI的发明者John Fenn和MALDI的发明者Koichi Tanaka在2002年一起分享了诺贝尔化学奖。下面将重点评述ESI离子源的最新进展。
1 ESI源的原理
1968年,Dole研究小组第一次使用了电喷雾离子源质谱分析了聚苯乙烯大分子[2]。他们发现聚苯乙烯在电离中会趋向于形成固定的带电物质,表明可以根据电喷雾离子源产生离子的质谱谱图来分析溶液中的物质,可以用作质谱中离子的来源。但由于无法获得稳定的质荷比以及对高电压的需求,这种实际上是分析大分子的离子化源的研究逐渐停止,直到1984年,Yamashita、Fenn和Alexandrov研究小组独立地研制出真正意义上的电喷雾离子源[3-4]。Fenn采用相对分子质量小于450的小分子作为分析物,缩短源和入口的距离,使得需要激发电子倍增器的电压减小,并与四极杆质谱分析器联用获得了负离子模式ESI源稳定的质谱图,从此ESI源正式成为质谱离子源的一员。但是此时能被检测的样品的相对分子质量还很小,直到1988年,Fenn研究小组才实现对多肽[5]和蛋白质[6]的检测,ESI源才真正成为生物大分子检测离子源。
ESI源的原理(以正离子模式为例):当强电场施加于喷雾针和取样孔后,溶液中负离子向喷雾头聚集,正离子向取样孔方向移动,形成泰勒(Taylor)锥。当Taylor锥表面的离子之间的静电排斥力达到溶液表面张力时,液滴会从喷雾头射出,在电场的作用下向取样口方向移动。当电荷密度和半径达到瑞利(Rayleigh)稳定限时,液滴在传输过程中由于溶剂蒸发,会因为库仑排斥作用发生分裂。Paul Kebarle和Liang Tang[7]的研究发现,液滴在达到瑞利稳定限的80%时就会发生分裂,且不是平均分裂成直径相同的小液滴,而是其中2%的质量被分裂出,并变成20个总共携带原液滴15%电荷的小液滴。如,当一个半径0.945μm携带51200个电荷的液滴分裂时,会产生1个半径0.939μm带43560个电荷的大液滴和20个半径0.09μm分别带384个电荷的小液滴。液滴会如此逐渐分裂,直到半径足够小并到达nm量级时,带电液滴会直接变成气相离子。气相离子的产生机制有两种解释,一种是由 Thomsona和lribarne[7-8]提出的离子蒸发模型(IEM),一种是 Dole[2,9]和 Röllgen[10]倡导的带电残渣模型(CRM),两种机制都有实验结果的支持。在IEM模型中,带电液滴表面的电荷密度随着溶剂的蒸发而增大,当带电液滴的小半径和高带电量满足瑞利稳定限时,液滴会分裂生成半径更小的液滴,这个过程反复进行直至半径降到8nm,带电量为70个电荷时,发生场助蒸发为止。高挥发性离子聚集在液滴表面,由于高电场作用,会直接从液滴中被弹出变为气相分子。CRM模型认为液滴经过逐渐的蒸发和分裂,当半径达到nm级时,待分析离子并不能移动到液滴表面发生场助蒸发,只能等待溶剂继续蒸发至完全,继承电荷形成一个带电的气相离子。一般认为,小分子离子由离子蒸发模型产生而带电,大蛋白气相离子的产生可由带电残渣模型解释[11-13]。两种模型中,待分析的离子在成为气相离子的过程中都不受到外界能量激发,不产生碎片,这种软电离的特性促进了ESI源在大分子质谱分析中的广泛使用。
2 影响ESI离子源取样效率的因素
在离子从产生到被检测器检测到的传输过程中,不可避免的在每个部分存在损失,传输效率可用来衡量该过程中每个部分损失的大小。诸如流速、电压和距离等离子源的一些设置,直接决定初始液滴的直径、动量和偏转方向,这就影响到有多少离子能进入取样口,如果离子偏转角度大,还会造成取样传输管内的损失,最终影响到喷雾针到取样口前甚至到截取锥前这部分的传输效率。而且离子源上安置不同的离子光学系统,对这部分的传输效率有直接影响。所以可以把这部分传输效率归于离子源性能的评价标准。它与源产生离子的效率即离子化效率一起构成的取样效率便是检验离子源性能的一个重要参数。一般认为,由于ESI源自身的特性可以产生带大量电荷的小液滴,所以源本身的离子化效率很高。1990年,Smith等[14]研究小组测得的细胞色素C的谱图证明ESI源本身离子化效率很高。当各种分析物以0.5~3μL/min速度通过毛细管时,他们测得离子以平均2×10-7A电流从喷雾针流出,只有平均2×10-9A电流到达取样孔,即传输效率为1%。当ESI源与一个四极杆质谱仪联用时,总离子中只有0.01%在被聚焦后进入四极杆中,只有0.001%的电流(即2×10-12A)最终到达检测器。细胞色素C的质谱谱图显示,平均每秒检测到的细胞色素C的离子数是3000个,而每秒进入ESI源的细胞色素C的物质的量为2.7×10-14mol,可计算得出细胞色素C离子的检测率为1.8×10-5,检测率的数量级和到达检测器电流的数量级一致,都为10-5,说明离子化效率很高,文献中假定的ESI源离子化效率为80%。
Kebarle等[15]给出了ESI源高离子化效率的理论解释。他们发现ESI源产生液滴的初始半径与流速的三分之二次方、溶液密度、溶液张力系数成正比。ESI源的喷嘴半径小,流速慢,使得液滴的初始半径很小,而同时带电量多,易分裂产生离子,并测得当分析物浓度小于10-3mol/L,流速为5μL/min时,初始产生的液滴半径约为1.5μm,而它的带电量为0.8×10-15C。当液滴的半径达到带电量对应的rayleigh稳定限半径的80%时,液滴会发生分裂,而对应这个带电量的分裂半径为0.945μm。根据表面蒸发限制法则,它只需要462μs的蒸发时间就达到这个半径,随着溶剂的蒸发不断分裂,再经过178μs时就会分裂成0.7μm的液滴,最终一共经过640μs就达到3nm半径的液滴。Smith等[14]测得在距离喷嘴0.5cm的地方液滴的直径已为0.3μm,说明在0.5cm左右的距离内离子化已经接近完成。现在的nanoESI源由于减少了流速使得液滴半径减小,可以达到几乎100%离子化效率。Schmidt等[16]用BaBr2检测了nanoESI源的离子化效率。BaBr2在正离子模式下可以被检测为Ba2+、Ba+、BaBr+,当初始液滴增大时,气相离子的产生就需要经过更多的溶剂蒸发过程,而趋向于形成BaBr+离子对,可用检测到的[Ba2++Ba+]和BaBr+离子量的比值来鉴定离子化效率。Schmidt发现当10-4mol/L的BaBr2溶液以30nL/s流速经过ESI源时,这个比值在1000以上,说明ESI的离子化效率接近于100%。
虽然ESI源的离子化效率很高,但传输效率很低,喷雾中只有1%的离子到达取样孔进入到真空系统中,说明传输效率才是制约ESI源取样效率的关键因素。
3 ESI源的改进
ESI源在离子喷雾理论的基础上分成了两个方向:低流速ESI源和高流速ESI源。
早在1994年,Caprioli[17]提出的 micro-ESI源和Wilm等[18]提出的nanoESI源即为最初的低流速ESI源。Caprioli采用的锥形尖头毛细管使流速减小到300~800nL/min。Wilm的拉伸包金毛细管使流速降到25nL/min。由于低流速ESI源流速的减小,与流速成正比的液滴半径也减小,需要蒸发的距离减小,因此通过减小ESI源到取样孔的距离,使得喷雾方式产生的大部分离子都可以进入取样孔中,传输效率得到了很大的提升。Wilm将包金拉长的玻璃毛细管放置在取样孔前1~2mm,当0.2μmol/L合成肽段溶液以22nL/min的流速进行检测时,检测率达到了1/390,是传统源检测率的510倍[19]。
由于低流速ESI源除了分辨率高,还具有上样量少的优点,因此采用低流速ESI源的液质联用分析促进了低流速液相色谱的研究,并发展成为今天普遍采用的nanoLC,这种小内径液相色谱已经成为了液质联用分析的重要组成部分。
但色谱学家有一种看法:样品的浓度是有限的,样品的体积是无限的,当用无限的体积去分析,总会得到分析结果,这也是对一些复杂的样品仍采用取样效率不高的高流速ESI源的原因[20]。
研究者们一直对高流速ESI源进行研究,目的是使得高流速液相色谱-质谱具有低流速液相色谱-质谱的高灵敏度。其方法是对离子喷雾过程进行改进,通过改善传输效率进而提高ESI源总的取样效率,主要的研究策略是通过对低流速源进行改进,并保证提高流速时不损害传输效率。增加取样口的内径确实可以提高传输效率,但同时也意味着需要更大的机械泵来保持真空状态,而且大口径的入口会造成原本的层状气流变的混乱和更多的离子损失,所以一般不采用[20-22]。通常提高传输效率的方法有两种:一种减小传输距离,使得喷雾离子化产生的大部分离子可以进入真空系统;另一种是对喷雾离子化产生的离子进行聚焦。
3.1 减小离子源与取样孔间的传输距离
离子源与取样孔间短的传输距离是低流速ESI源的特点,当流速提高,传输距离保持不变时,必然要损失离子化效率,所以这方面改进是对离子化效率损失的补偿。
代表性的工作是Schneider小组研制的离子选择接口PDI[23]。液滴从喷雾针喷出后进入一个长13mm,内径为1mm的可加热的圆柱形不锈钢腔室,腔室与一个内径为6mm,厚1mm的特氟龙圆环一起固定在取样板上,腔室末端形成的1mm厚的圆柱型薄层就构成了一个离子选择器,其结构示于图1。由于真空的吸力在腔室中会形成层状的气流,而层状的气流在到达真空入口后会弯曲,所以小离子会在选择器中随气流弯曲进入真空入口,而大离子会从气流中脱出,被反弹,再在强电场作用下向入口方向运动。这样对大离子而言就提高了溶剂蒸发时间,补偿了离子化效率。一般当将nanoESI源的流速提高到100~1000nL/min时会发生去溶剂化不足,检测到的离子数变弱的情况,但Schneider发现,当各种分析物以流速为500nL/min通过喷雾针时,采用PDI设计的ESI源平均离子数提高2倍,稳定性提高3倍,而且这种设计还对溶剂的组成,尤其是水溶液有很大的包容性,从而在nanoLC-MS联用时具有很高的稳定性[24]。
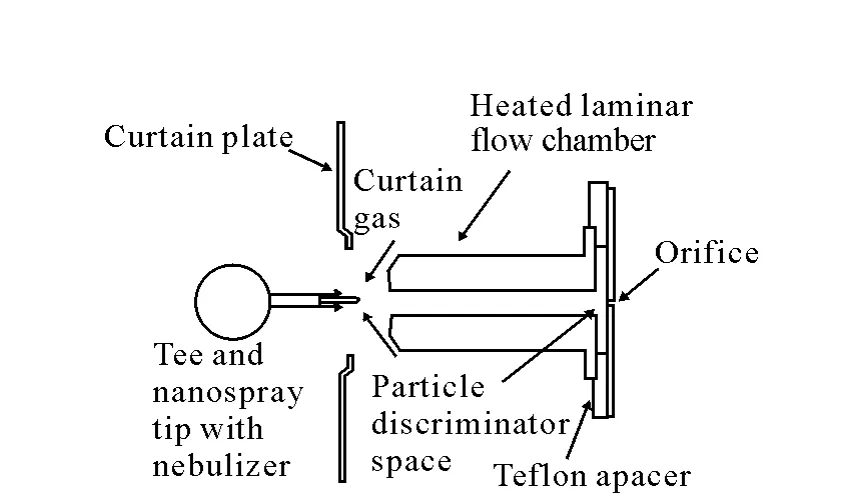
图1 粒子选择接口PDI原理图[23]Fig.1 Schematic of particle discriminator interface[23]
3.2 离子喷雾聚焦
离子喷雾聚焦主要有两种方法:一种是气动力学方法;另一种是电动力学方法。
气动力学代表的有Lee研究小组的文丘里管空气放大器[25],它的主要原理是文丘里效应和康达效应。文丘里效应是气流由粗变细通过文丘里管时会使气体在文氏管出口的后侧形成一个真空区。真空区会对附近的气体产生一定的吸附作用。康达效应是气流具有由本来的流动方向改为随着凸出的物体表面流动的行为。
当氮气进入环形气腔,再通过窄的环形间隙进入到空气放大器中,就产生文丘里效应,在空气放大器中央形成真空区,而氮气会由于康达效应随着管壁运动,从而拉伸液滴向气体前进方向移动,液滴在真空区也会加速去溶剂化,其原理示于 图 2。例 如,1μmol/L 利 血 平 以 1.5 μL/min的流速进行分析时,当文丘里管加直流电压而不加气流时,比原信号强度提高了0.5倍;当文丘里管加氮气而不加电压时,信号强度提高了5倍;当文丘里管既加氮气又加电压时,信号提高了18倍。Lee还发现这个提升是稳定的,不随分析物浓度变化而出现巨大波动。Yang等[26]实验证明这种气体放大器在对蛋白质分析时能实现温和的离子化,拥有更高的动态pH范围,且质谱峰由宽变窄。Robichaud等[27]通过压电传动装置精密控制环状带隙,研究带隙宽度和气压对放大器性能的影响,最终真空度获得2倍的提升,信号强度提高34倍。
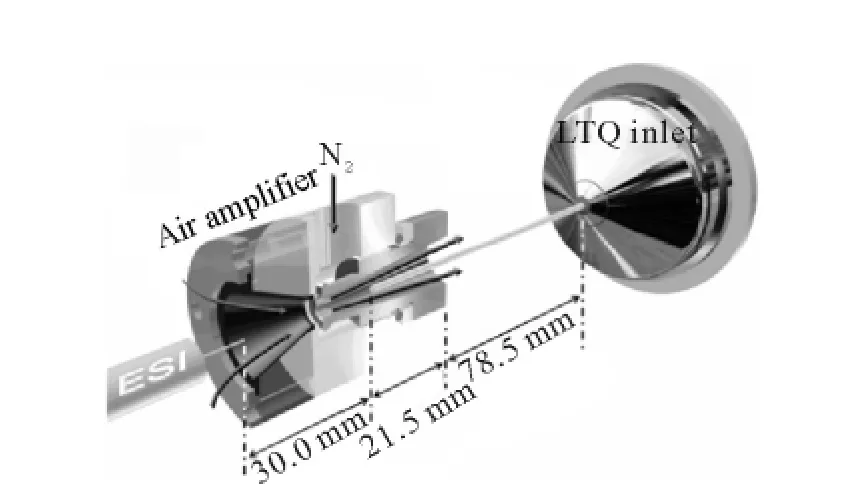
图2 ESI-AA-LTQ MS系统原理图[26]Fig.2 Schematic of the ESI-AA-LTQ MS system[26]
气动力学的另一个代表是Bruce等[28]用喇叭口取样管替代现有的直筒金属管,其结构示于图3。当内径0.38mm,外径0.76mm的金属管在末端向外展开至外径2.2mm后,安装在FT-ICR-MS上时,对10μmol/L的 P物质以250nL/min的流速进行分析,得到了2倍的离子强度和3倍的离子传输电流。同文丘里管原理相似,收缩的气流会减少离子云的扩散,使更多离子进入真空系统。Bruce还发现这种设计使得取样管对喷针位置的容忍性有很大提高,在轴线方向相距12mm时仍然具有80%的离子强度,而传统设计在5mm时就已减少了50%。这是由于气流在喇叭口的收缩作用帮助离子进入真空系统。相应的在垂直方向,离子源偏离1.2mm内没有明显的信号改变,而传统的离子源喷头在偏离最佳位置0.8mm时就几乎没有信号了。Dixon等[29]将喇叭口取样管和文丘里放大器联用,却没有发现大的信号上的改变,因此要从该设计获得更好的灵敏度还需要对实验条件进行优化。
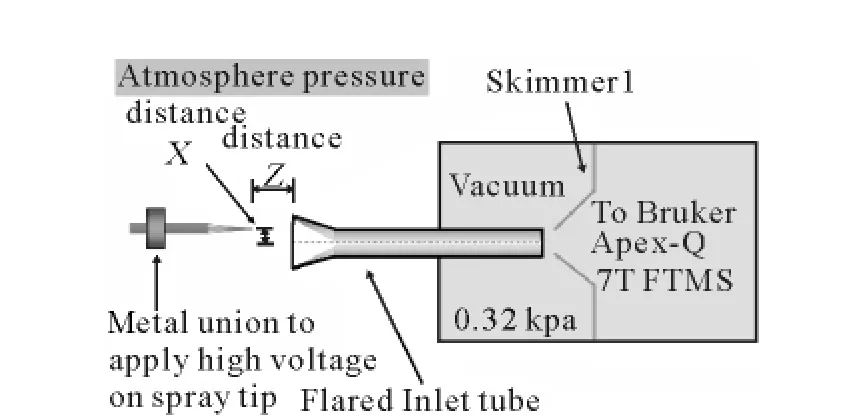
图3 喇叭口取样管示意图[28]Fig.3 Schematic of the implementation of the flared inlet tube[28]
气动力学虽然能有效地提升传输效率,但引入的中性粒子,一方面增加检测噪音,另一方面影响气相离子的传输效率。
电动力学方法不增加其他粒子,通过电力作用还可产生偏转排除中性粒子。电动力学方法又分为两种:射频电场和直流电场。
射频电场的代表是Richard研究小组的双重离子漏斗技术(subambient pressure ioniza-tion with nanoelectrospray,SPIN)[30],它来源于早期Richard的离子漏斗技术[31]。离子漏斗技术依据的原理是真空下RF mutipole技术可以通过碰撞实现离子的聚焦,是一种离子源和质谱真空系统的接口,可以提高真空系统的传输效率。离子漏斗是一组内径逐渐减小的镍包铜环,铜环外径为38mm,内径从22.2mm缩小到1mm,上接有从300V逐渐减小为50V的直流电压,相邻铜环接频率为700kHz,振幅为98V相位相反射频电压,因为聚焦要求真空环境,所以离子漏斗在1.3Pa下工作。采用三重四极杆质谱,在流速为400nL/min,分析浓度为4μmol/L马心细胞色素C时,传统ESI源获得的图谱TIC数为2.7×106,而使用了离子漏斗技术的ESI源获得图谱的TIC数为5.0×107,获得了18倍的提升。SPIN技术用离子漏斗来提升离子源的传输效率,其结构示于图4。将ESI源移入真空系统中,与离子漏斗直接相接,使用一个修改过的离子漏斗与一个传统的离子漏斗串联,ESI源处气压可提高到4kPa,并用50%甲醇作为溶剂进行分析,预示着液质联用的可行性。改进的离子漏斗通过削除过剩的材料,以减少总电容,并增加射频振幅和频率。Marginean等[32]引入CO2气体来解决低压环境下电击穿问题,并测得流速为50nL/min时,离子化效率达到50%。Tang等[33]用SPIN成功的与反向液相色谱联用,以流速100~400nL/min对胰酶肽段和完整细胞裂解液进行分析,获得了5~12倍信号的提升,提升来自于离子化效率的改善和传输效率。
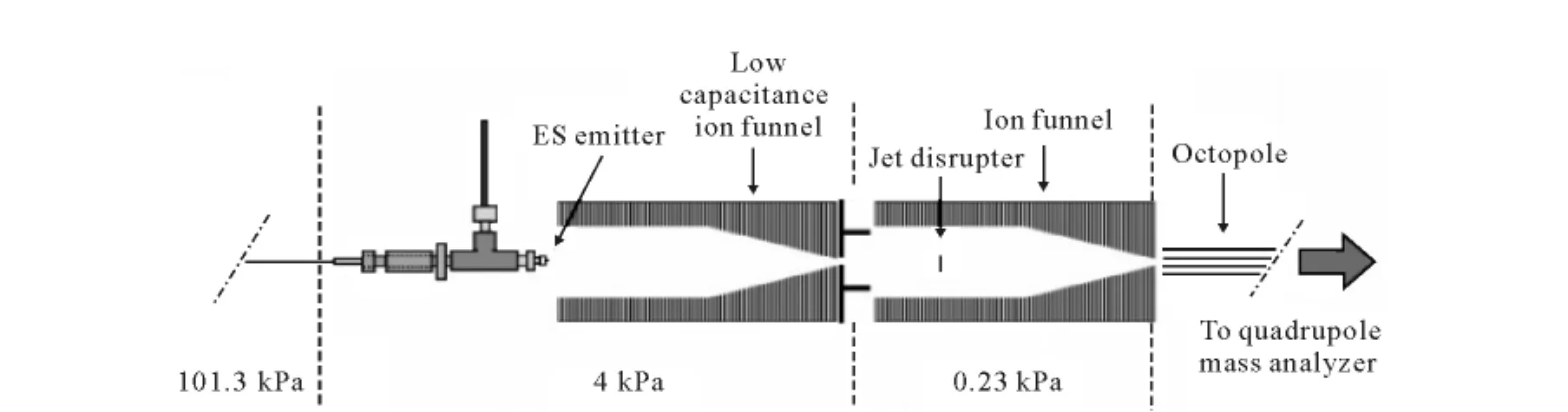
图4 SPIN 示意图[34]Fig.4 Schematic of subambient pressure ionization with nanoelectrospray source[34]
直流电场的代表是Schneider等[35]的离子透镜。喷雾针穿过一个1mm厚的不锈钢环,喷雾针的尖端超出不锈钢环2mm。不锈钢环上加载5100V电压时,产生的电场使得喷雾针到取样孔之间的电场线由原来发散状变为一起平行射向取样口,达到离子聚焦作用。以400 nL/min流速分析细胞色素C时,得到了3倍信号强度的提升。他们还发现不锈钢环加不同电压时,效果不同,当不锈钢环加电压为1600V,单电荷状态的峰m/z 1153最强,m/z 586的峰很小;当电压为2850V时,双电荷状态的峰m/z 586 最 强,m/z 1153 较 弱。Schneider等[36]根据这个效应设计了多重喷雾器,装置示于图5,2个喷雾器的不锈钢环上加不同电压,实现了利血平和细胞色素C信号强度在同一张谱内的一并提升。考察这种设计的非直射情况时,以1.5μL/min流速对缓激肽溶液进行分析,获得了1.5~4倍的提升,但背景噪音却无增加[37]。Zhong等[38]对离子透镜带来的电势变化采用有限元方法进行分析,并通过实验得到了与喷雾针电压相应的最优离子透镜电压。Zhao等[39]将离子透镜用于毛细管电泳质谱的ESI源中,成功扩大了毛细管电泳质谱稳定运行的流速和电压范围。

图5 装备离子透镜的4重电喷雾源[36]Fig.5 Photograph of a four-sprayer electrospray ion source with ion lens[36]
Thompson等[40]设计了另一种黄铜圆柱透镜,示于图6。直径为19mm,高为10.6mm的圆柱内部被加工成内凹的半球形,喷针从凹面最底处3.1mm直径的圆孔进入,距底面1mm处停止。并在距离喷针4mm的地方用自制3D离子电流测量装置,测得中心电流强度有5倍的增强,与原来相比,估计2倍的离子进入了四极杆质谱。
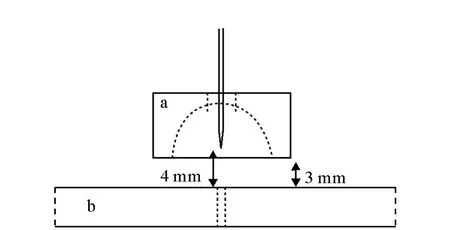
图6 Thompson和Jorgenson设计的离子透镜[40]Fig.6 The ion lens made by Thompson and Jorgenson[40]
3.3 对ESI源其他方面的改进
研究人员还根据不同的目的对ESI源的其他方面进行了改造,从而获得不同的效果。例如,为了减少肽段离子的电荷量,Zhang等[41]研究小组通过内径5mm,外径7mm的石英管包裹电喷雾针,并将电喷雾针上的直流电改为通过石英管上的锡纸加交流电使得液滴带电。改进前,Zhang测得醋酸强啡肽A(1-13)的3电荷峰强度最强,且电荷分布几乎不随直流电压增加而改变;改进后,当交流电压由3.5kV变为4.8 kV时,由3电荷峰强度最强变为双电荷峰强度最强,电压达到5.5kV时,单电荷峰强度最强。
研究者还将ESI源和其他离子化机制结合,研究出具有独特优势的源。如常温常压质谱(AMS)中的电喷雾解吸附离子化技术(DESI)是 Cooks[42-44]研究小组将 ESI源和解吸附离子化(DI)技术相结合的产物,该离子化源用ESI源产生的带电液滴或者初级离子喷射到分析物表面,萃取分析物后,溶剂蒸发,残留电荷使分析物变为带电气相离子。这种技术不需要或者只需很少步骤的样品预处理,可实现对样品的直接分析。Chen等[45-47]还在ESI源的基础上研制出三维空间的AMS技术——电喷雾萃取电离(EESI),通过ESI源产生带电的初级离子,在三维空间与样品喷雾器产生的中性液滴碰撞,通过萃取和电荷转移实现分析物的离子化。这种技术由于大的三维空间具有对分析物基体的容忍度,能对液体、胶体、气体进行直接分析,还可根据需要,通过产生不同的初级离子,选择性地萃取和离子化分析物,实现对其分析。类似的还有电喷雾辅助激光解吸电离(ELDI)[48-50]、激光消融电喷雾电离(LAESI)[51-52]、基质辅助激光解吸电喷雾(MALDESI)[53]、红外线激光辅助解吸电喷雾电离(IRLADESI)[54]、中性解吸电喷雾萃取电离(ND-EESI)[46]、纳升电喷雾萃取电离(nanoEESI)[55]、熔滴电喷雾萃取电离(FDESI)[56-57]等技术,这些技术中 ESI源或者作为初级离子产生装置,通过解吸萃取实现分析物的离子化,或者实现液态分析物的直接离子化。
对于这些离子化源来说,提高传输效率也极为重要,特别是源、分析物和质谱取样口三者之间存在一个较大的偏转角度时,待分析气相离子需要辅助外力进入到质谱中。
Abliz[58]研究小组的气流辅助离子化方法(AFAI)可与DESI离子化技术联用,装置示于图7,其特点是用泵将待分析离子吸入到传输管,到达传输管末端的离子由于惯性和质谱的真空吸力继续前进进入到取样口中。He等将AFAI技术与DESI源进行联用,对罗丹明6G进行分析,通过调节气流速度,获得了最大75倍信号强度的提升[58]。
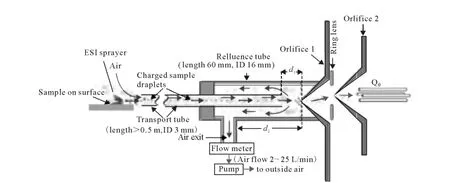
图7 气流辅助离子化装置(AFAI)示意图[58]Fig.7 Schematic of air flow assisted ionization(AFAI)[58]
Ouyang[59]研究小组用层状气流来解决角度歧视的问题。原来采用固定套管的DESI源直接与质谱取样口相接时,由于内径差距较大,相接位置前后两部分气体流速差距巨大,造成套管内样品表面存在强烈的涡流和分析物的损失[60]。离子传输装置示意图和模拟图示于图8。该研究小组采用中等内径传输管作为缓冲,减小了涡流强度,提升了离子的传输效率。而且传输管内产生的层状气流可以帮助离子进行长达6m的远距离传输。
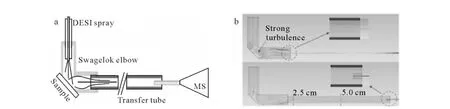
图8 Ouyang的离子传输装置示意图(a)和模拟图(b)[59]Fig.8 Schematic(a)and simulations(b)of Ouyang’s device for transferring ions[59]
4 展望
ESI源虽然由于自身的特点拥有超高离子化效率,但传输效率只有约1%,所以质谱学家通过对离子传输过程的改进就可以带来ESI源性能几十倍的提升,进而提高液相色谱与质谱联用分析的灵敏度和准确度,由此看出提高ESI源传输效率的研究依然是质谱研究领域的重点之一。另外,ESI源在常温常压条件下取样和直接取样的特点将会使ESI源质谱仪小型化、现场化和家庭化成为新的研究热点。
[1]MA X X,ZHANG S C,LIN Z Q,et al.Realtime monitoring of chemical reactions by mass spectrometry utilizing a low-temperature plasma probe[J].Analyst,2009,134(9):1863-1867.
[2]DOLE M,MACK L L,HINES R L,et al.Molecular beams of macroions [J].The Journal of Chemical Physics,1968,49(5):2240-2249.
[3]YAMASHITA M,FENN J B.Negative ion production with the electrospray ion source[J].Journal Name:J Phys Chem (United States),1984,88(20):4671-4675.
[4]ALEXANDROV M L,GALL L N,KRASNOV N V,et al.Extraction of ions from solutions under atmospheric pressure as a method for mass spectrometric analysis of bioorganic compounds[J].Rapid Communications in Mass Spectrometry,2008,22(3):267-270.
[5]WONG S F,MENG C K,FENN J B.Multiple charging in electrospray ionization of poly(ethylene glycols)[J].The Journal of Physical Chemistry,1988,92(2):546-550.
[6]MENG C K,MANN M,FENN J B.Of protons or proteins[J].Zeitschrift für Physik D Atoms,Molecules and Clusters,1988,10(2):361-368.
[7]IRIBARNE J V,THOMSON B A.Evaporation of small ions from charged droplets[J].Journal of Chemical Physics,1976,64(6):2287-2294.
[8]THOMSON B A,IRIBARNE J V.Fate of electrical charges in evaporating cloud droplets[J].Reviews of Geophysics,1978,16(3):431-434.
[9]MACK L L,KRALIK P,RHEUDE A,et al.Molecular beams of macroions.Ii[J].Journal of Chemical Physics,1970,52(10):4977-4986.
[10]SCHMELZEISEN R G,BÜTFERING L,RÖLLGEN F W.Desolvation of ions and molecules in thermospray mass spectrometry[J].International Journal of Mass Spectrometry and Ion Processes,1989,90(2):139-150.
[11]GAMERO-CASTANO M,de la MORA J F.Kinetics of small ion evaporation from the charge and mass distribution of multiply charged clusters in electrosprays[J].Journal of Mass Spectrome-try,2000,35(7):790-803.
[12]KEBARLE P,PESCHKE M.On the mechanisms by which the charged droplets produced by electrospray lead to gas phase ions[J].Analytica Chimica Acta,2000,406(1):11-35.
[13]FELITSYN N,PESCHKE M,KEBARLE P.Origin and number of charges observed on multiplyprotonated native proteins produced by esi[J].Int J Mass Spectrom,2002,219(1):39-62.
[14]SMITH R D,LOO J A,EDMONDS C G,et al.New developments in biochemical mass spectrometry:Electrospray ionization [J].Anal Chem,1990,62(9):882-899.
[15]KEBARLE P,TANG L.From ions in solution to ions in the gas phase-the mechanism of electrospray mass spectrometry[J].Analytical Chemistry,1993,65(22):972-986.
[16]SCHMIDT A,KARAS M,DULCKS T.Effect of different solution flow rates on analyte ion signals in nano-esi ms,or:When does esi turn into nano-esi[J].Journal of the American Society for Mass Spectrometry,2003,14(5):492-500.
[17]EMMETT M R,CAPRIOLI R M.Micro-electrospray mass spectrometry:Ultra-high-sensitivity analysis of peptides and proteins[J].Journal of the American Society for Mass Spectrometry,1994,5(7):605-613.
[18]WILM M S,MANN M.Electrospray and taylorcone theory,dole's beam of macromolecules at last?[J].International Journal of Mass Spectrometry and Ion Processes,1994,136(2/3):167-180.
[19]WILM M,MANN M.Analytical properties of the nanoelectrospray ion source [J]. Analytical Chemistry,1996,68(1):1-8.
[20]SCHNEIDER B B,JAVAHERI H,COVEY T R.Ion sampling effects under conditions of total solvent consumption[J].Rapid Communications in Mass Spectrometry,2006,20(10):1538-1544.
[21]BRUINS A P.Mass-spectrometry with ion sources operating at atmospheric-pressure[J].Mass Spectrometry Reviews,1991,10(1):53-77.
[22]KIM T,UDSETH H R,SMITH R D.Improved ion transmission from atmospheric pressure to high vacuum using a multicapillary inlet and electrodynamic ion funnel interface [J].Analytical Chemistry,2000,72(20):5014-5019.
[23]SCHNEIDER B B,BARANOV V I,JAVAHE-RI H,et al.Particle discriminator interface for nanoflow esi-ms[J].Journal of the American Society for Mass Spectrometry,2003,14(11):1236-1246.
[24]SCHNEIDER B,GUO X,FELL L,et al.Stable gradient nanoflow LC-MS[J].Journal of the A-merican Society for Mass Spectrometry,2005,16(9):1545-1551.
[25]ZHOU L,YUE B F,DEARDEN D V,et al.Incorporation of a venturi device in electrospray ionization[J].Analytical Chemistry,2003,75(21):5978-5983.
[26]YANG P X,COOKS R G,OUYANG Z,et al.Gentle protein ionization assisted by high-velocity gas flow [J].Analytical Chemistry,2005,77(19):6174-6183.
[27]ROBICHAUD G,DIXON R B,POTTURI A S,et al.Design,modeling,fabrication,and evaluation of the air amplifier for improved detection of biomolecules by electrospray ionization mass spectrometry[J].Int J Mass Spectrom,2011,300(2/3):99-107.
[28]WU S,ZHANG K,KAISER N K,et al.Incorporation of a flared inlet capillary tube on a fourier transform ion cyclotron resonance mass spectrometer[J].Journal of the American Society for Mass Spectrometry,2006,17(6):772-779.
[29]DIXON R B,MUDDIMAN D C.Quantitative comparison of a flared and a standard heated metal capillary inlet with a voltage-assisted air amplifier on an electrospray ionization linear ion trap mass spectrometer[J].Rapid Communications in Mass Spectrometry,2007,21(19):3207-3212.
[30]IBRAHIM Y,TANG K Q,TOLMACHEV A V,et al.Improving mass spectrometer sensitivity using a high-pressure electrodynamic ion funnel interface[J].Journal of the American Society for Mass Spectrometry,2006,17(9):1299-1305.
[31]SHAFFER S A,TANG K,ANDERSON G A,et al.A novel ion funnel for focusing ions at elevated pressure using electrospray ionization mass spectrometry [J].Rapid Communications in Mass Spectrometry,1997,11(16):1813-1817.
[32]MARGINEAN I,PAGE J S,TOLMACHEV A V,et al.Achieving 50%ionization efficiency in subambient pressure ionization with nanoelectrospray[J].Anal Chem,2010,82(22):9344-9349.
[33]TANG K,PAGE J S,MARGINEAN I,et al.Improving liquid chromatography-mass spectrometry sensitivity using a subambient pressure ionization with nanoelectrospray (spin)interface[J].J Am Soc Mass Spectrom,2011,22(8):1318-1325.
[34]PAGE J S,TANG K,KELLY R T,et al.Subambient pressure ionization with nanoelectrospray source and interface for improved sensitivity in mass spectrometry [J].Analytical Chemistry,2008,80(5):1800-1805.
[35]SCHNEIDER B B,DOUGLAS D J,CHEN D D Y.An atmospheric pressure ion lens to improve electrospray ionization at low solution flow-rates[J].Rapid Communications in Mass Spectrometry,2001,15(22):2168-2175.
[36]SCHNEIDER B B,DOUGLAS D J,CHEN D D Y.Multiple sprayer system for high-throughput electrospray ionization mass spectrometry [J].Rapid Communications in Mass Spectrometry,2002,16(20):1982-1990.
[37]SCHNEIDER B B,DOUGLAS D J,CHEN D D Y.An atmospheric pressure ion lens that improves nebulizer assisted electrospray ion sources[J].Journal of the American Society for Mass Spectrometry,2002,13(8):906-913.
[38]ZHONG X,YI R,HOLLIDAY A E,et al.Field distribution in an electrospray ionization source determined by finite element method[J].Rapid Communications in Mass Spectrometry,2009,23(5):689-697.
[39]ZHAO S S,ZHONG X F,CHEN D D Y.Atmospheric pressure ion lens extends the stable operational region of an electrospray ion source for capillary electrophoresis-mass spectrometry[J].Electrophoresis,2012,33(8):1322-1330.
[40]THOMPSON J W,ESCHELBACH J W,WILBURN R T,et al.Investigation of electrospray ionization and electrostatic focusing devices using a three-dimensional electrospray current density profiler[J].Journal of the American Society for Mass Spectrometry,2005,16(3):312-323.
[41]PENG Y E,ZHANG S,GONG X,et al.Controlling charge states of peptides through inductive electrospray ionization mass spectrometry[J].Analytical Chemistry,2011,83(23):8863-8866.
[42]TAKATS Z,WISEMAN J M,GOLOGAN B,et al.Mass spectrometry sampling under ambient conditions with desorption electrospray ionization[J].Science,2004,306(5695):471-473.
[43]TAKATS Z,WISEMAN J M,COOKS R G.Ambient mass spectrometry using desorption electrospray ionization(desi):Instrumentation,mechanisms and applications in forensics,chemistry,and biology[J].Journal of Mass Spectrometry,2005,40(10):1261-1275.
[44]COOKS R G,OUYANG Z,TAKATS Z,et al.Ambient mass spectrometry[J].Science,2006,311(5767):1566-1570.
[45]CHEN H W,VENTER A,COOKS R G.Extractive electrospray ionization for direct analysis of undiluted urine,milk and other complex mixtures without sample preparation [J].Chemical Communications,2006,(19):2042-2044.
[46]CHEN H W,YANG S,WORTMANN A,et al.Neutral desorption sampling of living objects for rapid analysis by extractive electrospray ionization mass spectrometry [J].Angew Chem-Int Edit,2007,46(40):7591-7594.
[47]CHEN H W,SUN Y P,WORTMANN A,et al.Differentiation of maturity and quality of fruit using noninvasive extractive electrospray ionization quadrupole time-of-flight mass spectrometry[J].Analytical Chemistry,2007,79(4):1447-1455.
[48]SHIEA J,HUANG M Z,HSU H J,et al.Electrospray-assisted laser desorption/ionization mass spectrometry for direct ambient analysis of solids[J].Rapid Communications in Mass Spectrometry,2005,19(24):3701-3704.
[49]CHENG C Y,YUAN CH,CHENG S C,et al.Electrospray-assisted laser desorption/ionization mass spectrometry for continuously monitoring the states of ongoing chemical reactions in organic or aqueous solution under ambient conditions[J].Analytical Chemistry,2008,80(20):7699-7705.
[50]PENG I X,SHIEA J,LOO R R O,et al.Electrospray-assisted laser desorption/ionization and tandem mass spectrometry of peptides and proteins[J].Rapid Communications in Mass Spectrometry,2007,21(16):2541-2546.
[51]NEMES P,VERTES A.Laser ablation electrospray ionization for atmospheric pressure,in vivo,and imaging mass spectrometry[J].Analytical Chemistry,2007,79(21):8098-8106.
[52]NEMES P,BARTON A A,LI Y,et al.Ambient molecular imaging and depth profiling of live tissue by infrared laser ablation electrospray ionization mass spectrometry[J].Analytical Chemistry,2008,80(12):4575-4582.
[53]SAMPSON J S,HAWKRIDGE A M,MUDDIMAN D C.Generation and detection of multiplycharged peptides and proteins by matrix-assisted laser desorption electrospray ionization(maldesi)fourier transform ion cyclotron resonance mass spectrometry[J].Journal of the American Society for Mass Spectrometry,2006,17(12):1712-1716.
[54]REZENOM Y H,DONG J,MURRAY K K.Infrared laser-assisted desorption electrospray ionization mass spectrometry [J].Analyst,2008,133(2):226-232.
[55]LI M,HU B,LI J Q,et al.Extractive electrospray ionization mass spectrometry toward in situ analysis without sample pretreatment[J].Analytical Chemistry,2009,81(18):7724-7731.
[56]CHANG D Y,LEE C C,SHIEA J.Detecting large biomolecules from high-salt solutions by fused-droplet electrospray ionization mass spectrometry[J].Analytical Chemistry,2002,74(11):2465-2469.
[57]SHIEH I F,LEE C Y,SHIEA J.Eliminating the interferences from tris buffer and sds in protein analysis by fused-droplet electrospray ionization mass spectrometry [J].Journal of Proteome Research,2005,4(2):606-612.
[58]HE J,TANG F,LUO Z,et al.Air flow assisted ionization for remote sampling of ambient mass spectrometry and its application[J].Raid Communications in Mass Spectrometry,2011,25(7):850.
[59]GARIMELLA S,XU W,HUANG G M,et al.Gas-flow assisted ion transfer for mass spectrometry[J].Journal of Mass Spectrometry,2012,47(2):201-207.
[60]VENTER A,COOKS R G.Desorption electrospray ionization in a small pressure-tight enclosure[J]. Analytical Chemistry,2007,79 (16):6398-6403.
猜你喜欢
节能技术(2022年4期)2022-11-08
核技术(2022年7期)2022-07-22
分析化学(2020年8期)2020-08-21
生物工程学报(2020年7期)2020-07-29
中南大学学报(自然科学版)(2019年7期)2019-08-13
科技视界(2019年18期)2019-08-07
中国新技术新产品(2019年23期)2019-01-20
分析化学(2018年4期)2018-11-02
分析化学(2018年7期)2018-09-17
分析化学(2018年7期)2018-09-17
