
落新妇苷固体分散体在大鼠体内的药动学研究
2013-03-06 08:29:07胡凯扈荣许鹏庆白海波傅旭春
中国现代应用药学 2013年10期
胡凯,扈荣,许鹏庆,白海波,傅旭春
(1.浙江大学药学院,杭州 310058;2.杭州华东医药集团生物工程研究所有限公司,杭州 310011;3.浙江大学城市学院药物研究所,杭州 310015)
落新妇苷固体分散体在大鼠体内的药动学研究
胡凯1,2,扈荣2,许鹏庆2,白海波2,傅旭春3*
(1.浙江大学药学院,杭州 310058;2.杭州华东医药集团生物工程研究所有限公司,杭州 310011;3.浙江大学城市学院药物研究所,杭州 310015)
目的 研究大鼠口服落新妇苷固体分散体后的药动学参数。方法 采用HPLC测定大鼠口服落新妇苷固体分散体后血浆中的落新妇苷血药浓度。用Kinetica软件计算药动学参数。结果 落新妇苷在大鼠体内的药动学过程符合二室模型。AUC0-480min为(1.98±0.60)mmol·min·L-1。结论 将落新妇苷制成固体分散体可以显著提高其生物利用度。
落新妇苷;固体分散体;药动学;生物利用度
落新妇苷(3-O-α-L-鼠李糖-5,7,3´,4´-4羟基二氢黄酮醇)存在于虎耳草科植物落新妇、百合科植物土茯苓、胡桃科植物黄杞、金粟兰科植物草珊瑚等多种植物中[1-4]。
落新妇苷具有抑制辅酶A还原酶、抑制醛糖还原酶、肝保护、抗水肿、抗氧化等活性[5],可用于治疗免疫系统或肝损伤等疾病。但落新妇苷难溶于水,口服吸收差,生物利用度低[6],是研究开发其在临床上应用所迫切需要解决的问题。本实验用溶剂法将落新妇苷制成固体分散体,研究了落新妇苷固体分散体大鼠灌胃给药的体内药动学性质,为落新妇苷的剂型研究和临床应用提供参考。
1 仪器与试剂
1.1 仪器
高效液相色谱仪(日本岛津,LC-20AT泵、SPD-20A检测器、SIL-20A自动进样器、SHIMADZU LCsolution化学工作站);VW-80A涡旋混合器(上海医大仪器厂);Allegra 64R Centrifuge高速离心机(BECKMAN COULTER公司)。
1.2 药品与试剂
落新妇苷(从黄杞叶中提取得到,纯度:95.5%,由杭州华东医药集团生物工程研究所提供,批号:100801);落新妇苷对照品(成都普思生物科技有限公司,批号:PS10013001,纯度>99%);甲醇(色谱纯,J&K SCIENTIFIC LTD,批号:LL30L01);磷酸(分析纯,国药集团化学试剂有限公司,批号:T20090304);磷酸二氢钾(分析纯,湖州湖试化学试剂有限公司,批号:100401);无水乙醇(分析纯,安徽安特生物化学有限公司,批号:1107073601);聚维酮PVP K30(上海昌为医药辅料技术有限公司,批号:05000244868);水合氯醛(上海化学试剂采购供应五联化工厂,批号:20100815)。
1.3 动物
SD大鼠,♂,体质量300~400 g,浙江省实验动物中心提供,许可证号:SCXK(浙)2008-0033。
2 方法
2.1 落新妇苷固体分散体的配制
称取落新妇苷适量,以PVP为载体,乙醇为溶剂,采用溶剂法制成固体分散体。临用前以水稀释,使落新妇苷含量为44.4 mmol·L-1。
2.2 色谱条件
Syncronis C18(150 mm×4.6 mm,5 μm)色谱柱;流动相为甲醇-0.01 mol·L-1KH2PO4-磷酸(30∶70∶0.1),流速1.0 mL·min-1,柱温35 ℃;检测波长291 nm。进样体积20 μL。
2.3 血浆样品的处理
取血浆100 μL,加入甲醇150 μL,静置沉淀,15 000 r·min-1离心10 min,吸取上清液进样分析。
2.4 动物实验
SD大鼠5只,禁食不禁水12 h,腹腔注射10%水合氯醛进行麻醉后,然后进行一侧颈动脉插管手术。以落新妇苷固体分散体的水溶液灌胃,灌胃剂量以落新妇苷计均为100 mg·kg-1。并于给药后5,10,15,20,25,30,60,120,180,240,300,360,480 min颈动脉取血0.3 mL,所得血样均按“2.3”项下方法处理,HPLC测定落新妇苷含量。
3 结果
3.1 专属性试验
空白血浆和落新妇苷血浆样品的HPLC色谱图见图1。落新妇苷色谱峰的保留时间为34.2 min左右,血浆中的内源性物质对落新妇苷的测定没有干扰。
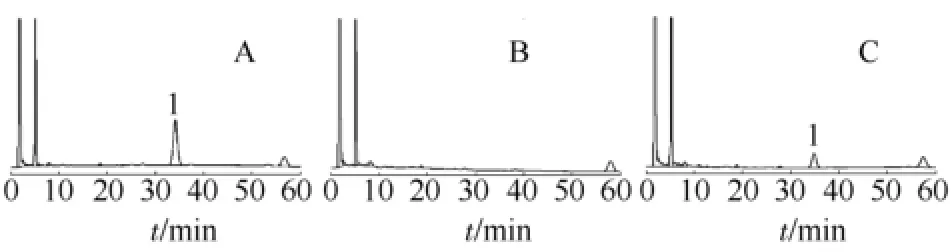
图1 落新妇苷大鼠血浆样品的HPLC色谱图A-对照品血浆样品;B-空白血浆;C-固体分散体灌胃给药后的血浆样品;1-落新妇苷Fig 1 HPLC chromatograms of rat plasma samples A-plasma samples of astilbin reference substances; B-blank plasma; C-plasma sample after oral administration of solid dispersion of astilbin; 1-astilbin
3.2 线性范围和检测限
在空白血浆中加入落新妇苷对照品溶液,配制浓度分别为0.07,0.14,0.28,0.55,1.10,2.20,4.39,8.78和17.56 μmol·L-1的血浆样品,按“2.3”项下方法操作,进行HPLC分析。以落新妇苷峰面积A对落新妇苷浓度C(μmol·L-1)进行线性回归,得落新妇苷的标准曲线方程为A=17 955.7C+ 3 916.8,r=0.998。在浓度0.07~17.56 μmol·L-1内,线性关系良好。最低定量限为0.07 μmol·L-1。
3.3 回收率与精密度
配制落新妇苷对照品血浆样品,使其浓度分别为0.09,2.38和17.56 μmol·L-1,按“2.3”项下方法操作,测定落新妇苷峰面积,并与相同浓度的落新妇苷对照品甲醇溶液直接进样得到的色谱峰面积进行比较,计算得到绝对回收率分别为81.2%,84.2%,86.4%(n=5)。实验还测定了落新妇苷浓度分别为0.09,2.38和17.56 μmol·L-1血浆样品的日内精密度和日间(3 d)精密度,日内精密度RSD分别为10.4%、6.0%和3.0%(n=5),日间(3 d)精密度RSD分别为10.1%,1.6% 和3.9%(n=5)。回收率与精密度均符合生物样品测定方法的要求。
3.4 动力学参数
所得血药浓度数据经Kinetica药动学软件进行拟合,结果表明落新妇苷固体分散体符合二室模型,其主要药动学参数见表1,平均血药浓度-时间曲线见图2。
表1 落新妇苷的药动学参数(n=5,±s )Tab 1 Phamarcokinetic parameters of astilbin(n=5,±s )
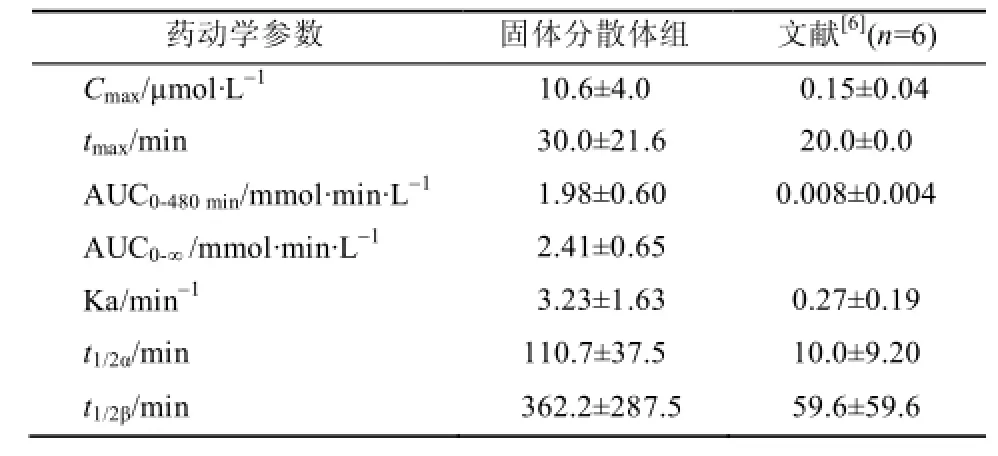
表1 落新妇苷的药动学参数(n=5,±s )Tab 1 Phamarcokinetic parameters of astilbin(n=5,±s )
AUC0-∞/mmol·min·L-12.41±0.65 Ka/min-13.23±1.63 0.27±0.19 t1/2α/min 110.7±37.5 10.0±9.20 t1/2β/min 362.2±287.5 59.6±59.6
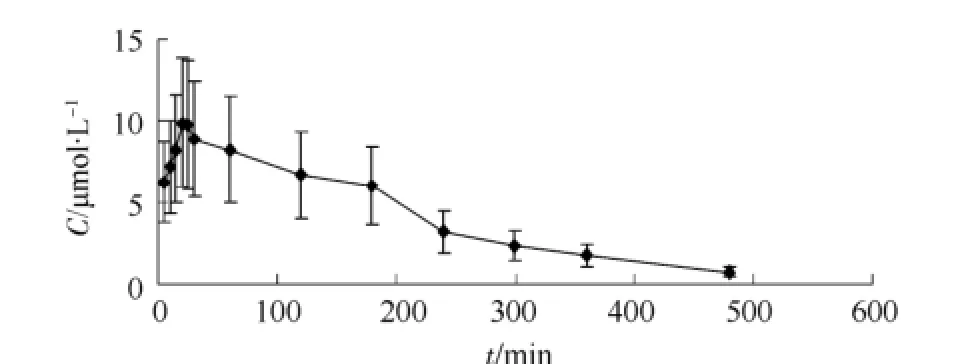
图2 落新妇苷的血药浓度-时间曲线Fig 2 Plasma concentration-time curve of astilbin
4 讨论
落新妇苷的口服吸收非常差,生物利用度低。有文献报道,用含0.886 mol·L-1乙醇的生理盐水配制的落新妇苷以100 mg·kg-1的剂量大鼠口服给药,UPLC-MS测定血药浓度,得AUC0-480min为(0.008±0.004)mmol·min·L-1[6]。
本实验测得落新妇苷在25 ℃时的溶解度只有1.1 mmol·L-1,落新妇苷的口服吸收差可能与其水溶性差有关。通过制剂技术提高难溶性药物的溶解度是改善其口服吸收的重要方法。
固体分散制剂是上世纪六十年代初才提出来的一种比较新的剂型,通常是将难溶性药物分散在水溶性载体中形成高度分散的固体分散体。固体分散体具有增大溶解度(本实验测得落新妇苷固体分散体在25 ℃时的溶解度>44.4 mmol·L-1)、加快溶出速率、提高生物利用度等优点,在改善难溶性中药有效成分的药物动力学性质方面的应用越来越广泛[7-12]。
从表1可以看出,落新妇苷制成固体分散体后,口服吸收情况明显改善。与生理盐水配制的落新妇苷相比,相对生物利用度为250。
进一步分析表1所列的2种剂型的落新妇苷大鼠口服给药的药动学参数,落新妇苷固体分散体的达峰时间有所延迟,吸收速率常数和峰浓度显著增大,分布速率和消除速率明显减缓,表明落新妇苷固体分散体不仅具有良好的吸收性,而且还具有明显的缓释性。
大鼠口服落新妇苷制剂的药动学研究的文献报道很少。本实验以颈动脉取血的方法研究了大鼠口服落新妇苷固体分散体后的体内药动学参数。研究结果表明:将落新妇苷制成固体分散体可以显著提高其生物利用度。
REFERENCES
[1] Nanjing University of Chinese Traditional Medicine. Dictionary of Chinese Traditional Medicine(中药大辞典) [M]. 2th ed. Shanghai: Shanghai People´s Publishing House, 1997: 3226.
[2] CHEN G Y, SHEN L S, JIANG P F. Study on the flavanonols glycosides in Smilacis glabra [J]. China J Chin Mater Med(中国中药杂志), 1996, 21(6): 355-357.
[3] QU J, ZHOU J, HOU W B, et al. Determination of astilbin and engelitin in engelhardtia leaf by HPLC [J]. Chin Tradit Herb Drugs(中草药), 2009, 40(2): 306-307.
[4] LI K Q, ZHANG G C, SHONG Y N, et al. Analyse astilbin and naringin in Sarcandra glabra by HPLC [J]. Chin Tradit Herb Drugs(中草药), 2010, 41(1): 137-138.
[5] GUO J M, XU Q, CHEN T. Quantitative determination of astilbin in rabbit plasma by liquid chromatography [J]. J Chromatography B, 2004, 805(2): 357-360.
[6] WANG X D. Study on the transport of taxifolin and astilbin in vitro and pharmacokinetics in rats [D]. Hangzhou: Zhejiang University, 2009.
[7] MA L Y, HAN L M, ZHANG Z R, et al. Preparation and characterization of solid dispersions of ginkgolides [J]. China J Chin Mater Med(中国中药杂志), 2009, 34(11): 1368-1372.
[8] LIU Y, CHANG J, HAN M H, et al. Preparation and in vitro release evaluation of isoniazid solid dispersion [J]. Chin J Mod Appl Pharm(中国现代应用药学), 2011, 28(2): 142-145.
[9] HAN G, YAN L Q, SUO W, et al. Preparation of emodin solid dispersion and determination of its dissolution [J]. Chin Tradit Herb Drugs(中草药), 2011, 42(3): 487-490.
[10] MA Y, LI W Z, GU J H. Preparation and evaluation of the solid dispersions of poorly soluble silybin [J]. J Chin Pharm Sci, 2011, 20(6): 604-608.
[11] WANG M R, WANG L Z, LIN J Y. Studies on preparation of candesartan solid dispersion and its dissolution and stability [J]. Chin J Mod Appl Pharm(中国现代应用药学), 2011, 28(7): 654-658.
[12] LIU X, WEN X G, LIAO X, et al. Progress in the study on physical stability and anti-aging of soild dispersion [J]. Chin J Mod Appl Pharm(中国现代应用药学), 2011, 28(8): 710-717.
Study on Pharmacokinetics of Astilbin Solid Dispersion in Rat
HU Kai1,2, HU Rong2, XU Pengqing2, BAI Haibo2, FU Xuchun3*
(1.College of Pharmaceutical Sciences, Zhejiang University, Hangzhou 310058, China; 2.Hangzhou Huadong Medicine Group Biotechnology R&D Institute Co., Ltd, Hangzhou 310011, China; 3.Institute of Materia Medica, Zhejiang University City College, Hangzhou 310015, China)
OBJECTIVE To study the pharmacokinetics of astilbin solid dispersion in rat. METHODS HPLC was used to determine the astilbin concentration in the plasma of rats. The pharmacokinetic parameters were calculated with Kinetica software. RESULTS After the rats were orally administrated with astilbin solide dispersions, the pharmacokinetic process of astilbin in rats was found to be consistent with the two-compartment model. The AUC0-480minwas (1.98±0.60)mmol·min·L-1. CONCLUSION The astilbin in solid dispersion can greatly improve its bioavailability in rat.
astilbin; solid dispersion; pharmacokinetics; bioavailability
R969.1
B
1007-7693(2013)10-1102-03
2013-03-04
胡凯,男,工程师 Tel: (0571)89918210 E-mail: hutingfish@sina.com*
傅旭春,男,博士,教授 Tel: (0571)88018711 E-mail: fuxc@zucc.edu.cn
猜你喜欢
中国现代医药杂志(2020年10期)2020-12-14 07:20:06
中成药(2019年12期)2020-01-04 02:02:24
成都大学学报(自然科学版)(2019年3期)2019-10-16 08:19:18
天津医科大学学报(2019年3期)2019-08-13 06:53:16
食品与发酵工业(2017年12期)2018-01-03 05:43:49
中成药(2017年10期)2017-11-16 00:49:54
中成药(2017年5期)2017-06-13 13:01:12
兽医导刊(2016年12期)2016-05-17 03:51:51
食品研究与开发(2015年8期)2015-10-27 02:00:18
现代检验医学杂志(2014年1期)2014-02-06 01:29:48
