
NC/NG共混体系的分子动力学模拟研究
2013-02-23 06:44齐晓飞张晓宏李吉祯刘芳莉宋振伟张军平
兵工学报 2013年1期
齐晓飞,张晓宏,李吉祯,刘芳莉,宋振伟,张军平
(1.西安近代化学研究所,陕西 西安710065;2.西北工业大学 理学院,陕西 西安710072)
0 引言
复合改性双基推进剂(CMDB)能量高、燃烧性能好,是战术导弹和火箭武器系统中常用的推进剂品种[1-2]。该类推进剂通过NG 增塑NC 所形成的物理交联网络将其他组分粘接在一起,并赋予推进剂一定的力学性能。因此,研究NG 与NC 的相互作用可以深入了解推进剂结构与性能的关系,为其综合性能的调节提供指导。虽然一些研究者通过扫描电镜观测[3-4]、拉伸实验[5-6]、动态扭辩分析[7-8]以及动态热机械分析[9-10]等方法得到了NG 与NC 相互作用的表观映像和定性分析结论,但由于实验尺度限制,对于他们之间相互作用的细节等理论机理方面,缺乏更为深入的了解。而分子动力学(MD)模拟方法作为近年来发展较为迅速的微尺度数值模拟方法,因其能够在分子水平上研究材料的微观结构并揭示组分之间的关系[11-13],已成为研究固体推进剂宏观性质本质的一个强力手段。付一政等[14]采用分子动力学研究了HTPB 推进剂中粘结剂与不同种类增塑剂以及铝粉之间的相互作用本质;张丽娜等[15]则研究了GAP 推进剂中粘结剂与固体填料RDX、HMX 和AP 间的相互作用,其结论与力学性能实验结果相吻合;但至今未见MD 模拟方法应用于CMDB 推进剂结构与性能研究的报道。
本文通过建立NC 纯物质、NC/NG 共混体系的微观结构模型并进行MD 模拟,在分子水平上认识NC 与NG 间相互作用的实质,为探索结构更复杂的CMDB 推进剂的宏观性质本质奠定基础,同时也为研究者从根本上提高该类推进剂性能提供一定的理论依据和参考。
1 MD 模拟
1.1 物理建模过程
依据NC 和NG 化学结构式,采用Materials Studio 4.0 的Visualizer 模块,建立了NC 和NG 的分子物理模型,如图1所示。考虑到模拟体系的分子大小对模拟效果的影响[16]和模拟效率因素,所建立的NC 分子链由20 个聚合单元组成,并以羟基封端。NC 分子链上的—OH 基团和—NO2基团的数目基于NC 含氮量(11.87%)计算得到,并随机分布于分子链上。为避免分子模型的能量陷入势阱,先采用COMPASS 力场[17],利用Smart Minimizer 方法对其进行能量优化。然后利用Amorphous Cell 模块的Construction 构建含有两个分子链的NC 纯物质模型(将两条NC 分子链的初始构象分别标记为NC-A和NC-B),然后向其中分别添加30 个、60 个和90个NG 分子,构建不同质量比NC/NG 共混体系模型(2NC 及2NC/30NG 模型见图1,其余略),其中NC的含量分别为75.06%、60.05%和50.04%.

图1 NC、NG 及其共混体系模型Fig.1 Models of NC,NG and their amorphous cell
1.2 MD 模拟细节
首先利用Smart Minimizer 方法对所构建的无定形分子模型进行能量优化,然后选用COMPASS 力场;应用周期性边界条件,即以立方元胞为中心,周围有26 个相邻的镜像立方元胞,以达到利用较少分子模拟宏观性质的目的。各分子起始速度由Maxwell-Boltzmann 随机分布给定,Velocity Verlet 算法进行求解。对分子间的范德华和静电作用力计算分别采用Atom-based 和Ewald 方法,非键截取半径0.95 nm,样条宽度取0.1 nm,缓冲宽度取0.05 nm.在293 K、303 K、313 K 和323 K 及1.01 ×105Pa 条件下,采用Andersen 控温方法和Berendsen 控压方法利用discover 模块进行400 ps、时间步长为1 fs 的NPT(正则系综,系统的粒子数N、压强P 和温度T 恒定)MD模拟,每100 fs 取样一次,记录模拟轨迹。后200 ps体系已经平衡(温度和能量随时间的变化率小于5%),对其MD 轨迹进行分析来获取回转半径和径向分布函数。
2 结果与讨论
2.1 NC 分子的回旋半径分析
回转半径指线性聚合物分子链中每个链节与分子链质心之间距离的统计平均值,是能够直接反映线性分子链构象的特征参数。由于NG 与NC 分子间的相互作用必然会影响NC 分子链的构象,同时考虑到温度也是决定NC 分子链构象的一个重要因素,因此通过回转半径来研究NC 分子链构象受NG分子数量和温度影响的变化,进而得到NG 与NC 分子间相互作用的直观映像。NC 纯物质及不同质量比的NC/NG 共混体系模型中,两条NC 分子链(NCA 和NC-B)在不同温度下的回转半径见表1.
CMDB 推进剂制备过程中的温度控制范围一般为293~323 K,由表1中数据可知,在此温度区间内随着温度的升高,各模型中NC 分子链的回转半径逐渐增加,但增加的趋势并不明显,即无论是否加入NG 分子,以及NG 分子数量的多寡,升高温度对NC分子链构象的影响并不显著。一般认为,升高温度会使高分子链段运动加剧,导致高分子链尺寸增大,回转半径增加;且分子量越大回转半径增加得越显著,相反,分子量越小回转半径增加得越平缓[18]。而对于本文中各模型,由于考虑到机时等模拟效率问题,NC 分子链的链节数量只有40,相对实际NC分子量较小,使得NC 分子链的回转半径随温度升高而增加的趋势并不明显,但这也表明了本文的模型能够准确反映NC 分子链构象受温度影响的变化。同由表1中数据可知,相对于温度因素,NG 分子数量对NC 分子链回转半径的影响较大,且NG 分子数量越多回转半径增加的越显著。如NG 分子由60 个增加至90 个时,NC-A 的回转半径由1.9 nm左右跃升至2.6 nm 左右,增大了36.8%;NC-B 由1.7 nm 左右跃升至2.3 nm 左右,增大了35.3%,二者回转半径的增幅均非常显著。
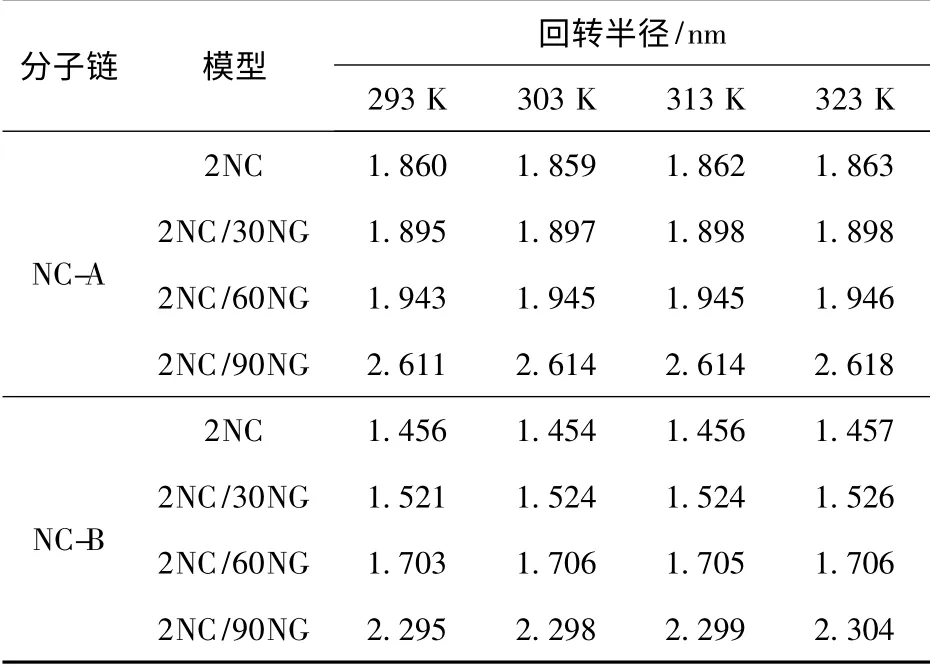
表1 各模型中NC 分子链在不同温度下的回转半径Tab.1 Radius of gyration for NC at different models and temperatures
NC 是由葡萄糖酐环状残基组成的线性聚合物,具有一定的刚性,但其环间的醚链又使链节的内旋转比较容易,所以NC 分子链具有一定的内旋自由度,但这种内旋自由度会受到NC 分子链中各原子间作用力的抑制。因此,推断上述NC 分子链回转半径增加的原因,是由于升高温度以及NG 分子的加入对NC 分子链内部的作用力均有弱化作用,使其内旋自由度增大,NC 分子链尺寸增大所致。但这一推论仍需进一步的研究加以证实,对此将在后续部分进行详细论述。
2.2 径向分布函数分析
径向分布函数g(r)是反映材料微观结构的特征物理量,它表示在距离某一设定中心粒子A 为r处,另一设定粒子B 的数目密度与B 的平均数目密度的比值,即
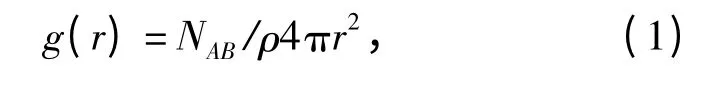
式中:NAB表示与中心粒子A 距离为r 到r +dr 处粒子B 数目;ρ 表示粒子B 的平均数目密度。
因此,可以通过分析径向分布函数中峰的位置来判断相互作用力的类型,并根据峰值的高低推断作用力的强弱。分子间存在的非键合力主要可分为氢键作用力和范德华力,前者的作用范围为0.26~0.31 nm,后者的作用范围为0.31~0.50 nm.由于氢键作用力的强度远大于范德华力,且考虑到温度变化对本文模型中NC 分子链构象的影响相对较小,因此只对303 K 下,各模型中能够形成氢键作用原子对的径向分布函数进行了模拟计算,从氢键作用力的角度分析NC 分子链回转半径变化的原因。
2.2.1 NC 分子内径向分布函数分析
氢键的形成需要氢原子的参与,即氢原子在与负电性很大的原子X 以共价键结合的同时,还同另一个负电性大的原子Y 形成一个弱键,形式为X—H…Y.因此,NC 纯物质模型中,NC 分子内能够与—OH 基团中氧原子O1 形成氢键的原子有主链氧原子O2、—NO2基团中氧原子O3 和氮原子N1 以及与—NO2基团相连的氧原子O4,4 种原子对及其径向分布函数如图2所示。
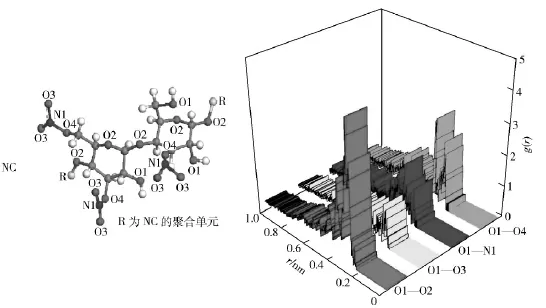
图2 2NC 模型中各原子对径向分布函数Fig.2 Radial distribution functions for different atoms at 2NC model
从图2可以看出,O1—O2、O1—O3 和O1—O4原子对在0.29 nm 附近出现峰,其中O1—O2 原子对的峰值最大,g(r)值为4.42;表明在这3 种原子对之间均形成了O—H…O 型氢键,且—OH 基团与主链上氧原子O2 之间的氢键作用最强。同时,4 种原子对在0.31~0.50 nm 范围内均出现了高低不同的峰,其中O1—O4 原子对的峰值最大,g(r)值为2.65;表明各原子对间存在强弱不一的范德华力,且—OH 基团与—ONO2基团上氧原子O4 之间的范德华力最强。上述模拟结果不仅证明了NC 分子链内部确实存在相互作用力,而且根据相互作用力的种类及其强弱,推断—OH 基团与主链上氧原子之间形成的氢键,可能是抑制NC 分子链内旋自由度的主要因素之一。
为了解NG 分子对NC 分子链内部相互作用力的影响,以氢键作用力最强的O1—O2 原子对为例,对它们之间径向分布函数随NG 分子增加而变化的趋势进行了分析,其结果如图3所示。由图3可知,随着NG 分子的增多,4 种模型中O1—O2 原子对径向分布函数曲线中的氢键峰值依次降低,分别为4.42、3.08、2.43 和2.11;此外,范德华力峰值也由2.34 依次降低至2.18、2.03 和1.56.这表明无论是—OH 基团与主链氧原子之间的氢键作用力还是范德华力,均随NG 分子的加入而减弱。即NG分子对NC 分子链内部的作用力有弱化作用,且随着NG 分子数量的增加弱化作用增强。
2.2.2 NG 与NC 分子间径向分布函数分析
在NG 分子中,能与NC 中—OH 基团中氧原子O1 形成氢键的有—NO2基团中的氧原子O5 和氮原子N2、与—NO2基团相连的氧原子O6.为找出NG分子使NC 分子链内部作用力弱化的原因,对以上3 种原子对O1—O5、O1—N2 和O1—O6 在2NC/90NG 模型中的径向分布函数进行了分析,其结果如图4所示。
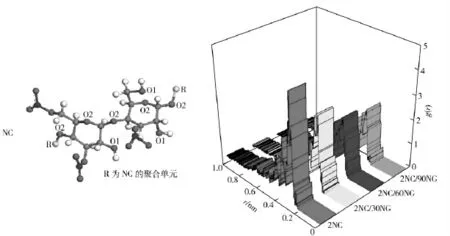
图3 不同模型中O1—O2 原子对径向分布函数Fig.3 Radial distribution functions for O1—O2 at different models

图4 2NC/90NG 模型中各原子对径向分布函数Fig.4 Radial distribution functions for different atoms at 2NC/90NG model
由图4可知,O1—O5 原子对与O1—O6 原子对可形成氢键,且前者的作用力更强,但后者的范德华力更强;而氧原子O1 与氮原子N2 之间只存在范德华力作用。这说明在NG 分子的—ONO2基团与NC分子的—OH 基团相互作用时,顶端氧原子O5 距离—OH 基团最近,氧原子O6 次之,氮原子N2 最远,即—ONO2基团倾向存在于—OH 基团的侧方。
图5为3 种NC/NG 共混物模型中O1—O5 原子对的径向分布函数曲线图,从中可看出,随着NG分子数的增加,O1—O5 原子对间的氢键作用力有所减弱,g(r)值由1.42 依次下降至1.34 和1.30.这是由于氢键具有饱和性,NG 分子数量增加后,单体NG 分子与NC 分子间形成的氢键数量减少;但由于NG 分子总数增加,总体上NG 与NC 分子之间的氢键数量增多,其相互作用力增强。
上述径向分布函数模拟结果表明,在NG/NC的共混体系中,NG 分子中—ONO2基团可与NC 分子—OH 基团形成氢键,从而替代NC 分子链之间的氢键,使其氢键作用力减弱,这可能是NG 分子加入后NC 分子链回转半径增大的原因。

图5 不同模型中O1—O5 原子对径向分布函数Fig.5 Radial distribution functions for O1—O5 at different models
3 结论
通过对NC 纯物质以及3 种不同质量比NC/NG共混体系的MD 模拟,并对其运动轨迹进行分析,得出以下结论:
1)在各模型中,升高温度对NC 分子链构象的影响并不显著;而NG 分子数量对NC 分子链回转半径的影响较大,且NG 分子数量越多回转半径增加的越显著。
2)NC 分子内存在较强的氢键作用力和范德华力,且这两种作用力随着NG 数量的增多而减弱。
3)NG/NC 的共混体系中,NG 分子中—ONO2基团可与NC 分子—OH 基团形成氢键,其强度随着NG 数量的增多而增强。
4)NC 分子内的氢键被NG 与NC 分子间的氢键所替代,可能是NG 分子加入后NC 分子链回转半径增大的原因。
References)
[1] 李上文,赵凤起,袁潮,等.国外固体推进剂研究与开发的趋势[J].固体火箭技术,2002,25(2):36 -42.LI Shang-wen,ZHAO Feng-qi,YUAN Chao,et al.Tendency of research and development for overseas solid propellants[J].Journal of Solid Rocket Technology,2002,25(2):36 -42.(in Chinese)
[2] 张海燕.改性双基低特征信号推进剂研究进展[J].固体火箭技术,2000,23(2):36 -38,43.ZHANG Hai-yan.Advances in low signature signal CMDB propellants[J].Journal of Solid Rocket Technology,2000,23(2):36-38,43.(in Chinese)
[3] 李吉祯,樊学忠,钟雷,等.NC/NG/AP/Al 复合改性双基推进剂力学性能研究[J].含能材料,2007,15(4):345 -348.LI Ji-zhen,FAN Xue-zhong,ZHONG Lei,et al.Mechanical properties of NC/NG/AP/Al composite modified double-base propellant[J].Chinese Journal of Energetic Materials,2007,15(4):345 -348.(in Chinese)
[4] 王瑛,张晓宏,陈雪莉,等.改性双基推进剂组合装药界面力学性能[J].含能材料,2011,19(3):287 -290.WANG Ying,ZHANG Xiao-hong,CHEN Xue-li,et al.Interfacial mechanical properties of single-chamber dual thrust grain for modified double-based propellant[J].Chinese Journal of Energetic Materials,2011,19(3):287 -290.(in Chinese)
[5] 王晗,樊学忠,刘小刚,等.浇铸型高能CMDB 推进剂的力学性能[J].含能材料,2010,18(1):88 -92.WANG Han,FAN Xue-zhong,LIU Xiao-gang,et al.Mechanical properties of casting high energy composite modified double-base propellant[J].Chinese Journal of Energetic Materials,2010,18(1):88 -92.(in Chinese)
[6] 邵重斌,付小龙,吴淑新,等.辅助增塑剂对AP-CMDB 推进剂力学性能的影响[J].化学推进剂与高分子材料,2010,8(6):50 -51,67.SHAO Chong-bin,FU Xiao-long,WU Shu-xin,et al.Influence of assistant plasticizers on mechanical properties of AP-CMDB propellants[J].Chemical Propellants & Polymeric Materials,2010,8(6):50 -51,67.(in Chinese)
[7] 贾展宁,周起槐.硝化纤维素、双基粘合剂和改性双基推进剂动态粘弹性分析[J].北京工业学院学报,1984,(3):72 -80.JIA Zhan-ning,ZHOU Qi-huai.Dynamic viscoelastic properties of nitrocellulose,DB and CMDB propellant[J].Journal of Beijing Institute of Technology,1984,(3):72 -80.(in Chinese)
[8] 贾展宁.改性双基推进剂力学性能特点分析[J].兵工学报,1989,10(3):17 -23.JIA Zhan-ning.Analysis of mechanical properties of CMDB propellants[J].Acta Armamentarii,1989,10(3):17 -23.(in Chinese)
[9] 刘子如,张腊莹,衡淑云,等.双基推进剂的玻璃化温度[J].火炸药学报,2009,32(2):56 -59.LIU Zi-ru,ZHANG La-ying,HENG Shu-yun,et al.The glass transition temperature for double base propellant[J].Chinese Journal of Explosives & Propellants,2009,32(2):56 -59.(in Chinese)
[10] 刘子如,张沛.增塑剂对双基推进剂动态力学性能的影响[J].固体火箭技术,2005,28(4):276 -279.LIU Zi-ru,ZHANG Pei.Influence of plasticizers on dynamic mechanical properties of double base propellants[J].Journal of Solid Rocket Technology,2005,28(4):276 -279.(in Chinese)
[11] 朱伟平.分子模拟技术在高分子领域的应用[J].塑料科技,2002,(5):23 -25,33.ZHU Wei-ping.Application of molecular simulation technology to macromolecule[J].Plastics Science and Technology,2002,(5):23 -25,33.(in Chinese)
[12] Matsuda T,Tai K.Computer simulation of the fracture of perfectly oriented polymer fibers[J].Polymer,1997,38(7):669 -676.
[13] Pan R,Liu X,Zhang A,et al.Molecular simulation on structure-property relationship of polyimides with methylene spacing groups in biphenyl side chain[J].Computation Materials Science,2007,39(4):887 -895.
[14] 付一政,胡双启,兰艳花,等.HTPB/增塑剂玻璃化转变温度及力学性能的分子动力学模拟[J].化学学报,2010,68(8):809 -813.FU Yi-zheng,HU Shuang-qi,LAN Yan-hua,et al.Molecular dynamics simulation on the glass transition temperature and mechanical properties of HTPB/Plasticizer blends[J].Acta Chimica Sinica,2010,68(8):809 -813.(in Chinese)
[15] 张丽娜,李定华,姚维尚,等.GAP 接枝海因与推进剂组分相互作用的分子模拟[J].推进技术,2010,31(5):587 -592.ZHANG Li-na,LI Ding-hua,YAO Wei-shang,et al.Molecular dynamics simulation of interaction between GAP grafted hydantoin and solid oxidizers in for GAP propellant[J].Journal of Propulsion Technology,2010,31(5):587 -592.(in Chinese)
[16] Brown D,Clarke J H R.Molecular dynamics simulation of an amorphous polymer under tension.1.Phenomenology[J].Macromolecules,1991,24(8):2075 -2082.
[17] Sun H,Ren P,Fried J R.The COMPASS force field:parameterization and validation for hosphazenes[J].Computational and Theoretical Polymer Science,1998,8(1 -2):229 -246.
[18] Rubinstein M,Colby R.Polymer physics[M].Oxford:Oxford University Press,2002.
猜你喜欢
椰城(2021年12期)2021-12-10
大连民族大学学报(2021年5期)2021-11-15
少儿科学周刊·少年版(2021年22期)2021-01-17
新课程·下旬(2019年7期)2019-09-17
发明与创新·中学生(2017年11期)2017-12-07
腐蚀与防护(2016年7期)2016-09-14
中学生数理化·高三版(2016年2期)2016-09-10
中学生数理化·高二版(2016年6期)2016-05-14
科学中国人(2016年35期)2016-01-28
新高考·高一物理(2015年11期)2015-12-24
