
2-偕二硝甲基-5-硝基四唑的合成、热性能及量子化学研究
2013-01-29 07:33葛忠学毕福强王伯周
火炸药学报 2013年3期
张 敏,葛忠学,毕福强,许 诚,刘 庆,王伯周
(西安近代化学研究所,陕西 西安710065)
引 言
唑类富氮化合物结构中存在较多的C-N 和N-N 键,具有较高的生成焓、较大的密度以及较好的氧平衡值,同时具备高气体生成量、低感度以及爆轰产物清洁等优点[1-2]。在唑环N 原子上引入偕二硝甲基得到N-偕二硝甲基唑类化合物,结构中存在偕二硝甲基和唑环单元,具有致爆基团多、能量适中、爆炸产物清洁等优点;偕二硝甲基单元使得N-偕二硝甲基唑类化合物具有较好的酸性和反应活性,易于衍生化,可作为构筑含能材料的有效单元。近年来有关N-偕二硝甲基唑类化合物的合成和性能研究受到国外相关人员的关注[3-5]。Semenov等人[3]合成了2-偕二硝甲基-5-硝基四唑(HDNMNT),氮质量分数为44.76%,氧平衡为10.96%。该研究通过溴丙酮与5-硝基四唑钠盐反应将丙酮基团引入四唑环的N2位,但由于溴丙酮难以购买,且毒性较大,因此该方法不易实施。
本研究用氯丙酮代替溴丙酮,以5-氨基四唑为原料,经过重氮化-取代反应、取代反应、硝化-水解反应三步合成了HDNMNT,实验过程更为安全环保,优化了关键前体2-丙酮基-5-硝基四唑(ANT)的合成工艺,研究了HDNMNT 的热性能,为其更深入研究提供参考。
1 实 验
1.1 仪器与试剂
德国Elementar公司Vario EL III型有机元素分析仪;美国Nicolet公司Nexus870型红外光谱仪;瑞士Bruker公司AV 500型(500MHz)超导核磁共振波谱仪;美国TA 公司901s差示扫描量热仪。
5-氨基四唑、亚硝酸钠、氯丙酮、浓硫酸、浓硝酸、丙酮、N,N-二甲基甲酰胺(DMF)、乙酸乙酯、乙腈和无水硫酸镁等均为分析纯,KBr为化学纯。
1.2 二水合5-硝基四唑钠盐(NaNT·2H2O)的合成
将5.150g(50mmol)5-氨基四唑溶于80mL水中,滴加1.216g 浓硫酸,升温至50℃,备用。将12.545g(180mmol)亚硝酸钠加入反应瓶中,用80mL水溶解。将上述5-氨基四唑-浓硫酸-水溶液缓慢滴加至亚硝酸钠溶液中,待体系稳定后,升温至60~70℃,反应2h。加入2.169g(54.2mmol)氢氧化钠,搅拌至溶解,旋蒸,冷却后加入丙酮提取,过滤,滤液用无水硫酸钠干燥,过滤,旋蒸,得到淡黄色液体转移至表面皿干燥得到6.66g淡黄色结晶固体,收率76.99% 。
13C NMR(DMSO-d6,125MHz):169.1;IR(KBr),υ(cm-1):3 411,1 687,1 552,1 454,1 319,1 417,841;元素分析(CH4N5NaO4,%):理论值,C 6.94,H 2.33,N 40.47;实测值,C 6.96,H 2.36,N 40.42。
1.3 2-丙酮基-5-硝基四唑(ANT)的合成
向25mL 三口烧瓶中加入5.190g(30mmol)NaNT·2H2O,用10mL DMF 溶解,加入5.358g(45mmol)KBr,再加入4.248g(45mmol)氯丙酮。升温至90℃,反应20min,倒入盛有100mL 水的烧杯中搅拌,过滤,即得到3.797g白色纤维状固体,收率74.02%。
1H NMR(DMSO-d6,500 MHz):6.17(s,2H,-CH2),3.32(s,3H,-CH3);13C NMR(DMSO-d6,125MHz):198.67(CO),166.01(C5),62.66(CH2),27.16(CH3);IR(KBr),υ(cm-1):3 001,2 954,1 733,1 578,1 420,1 340,1 315,1 180,1 074,848,578;元素分析(C4H5N5O3,%):理论值,C 28.05,H 2.93,N 40.91;实测值,C 28.08,H 2.95,N 40.93。
1.4 2-偕二硝甲基-5-硝基四唑(HDNMNT)的合成
向25mL三口烧瓶中加入1.515g(8.86mmol)2-丙酮基-5-硝基四唑(ANT)和7.2mL 浓硫酸,冰盐浴下滴加5.4mL 65%(质量分数)的硝酸,控制温度小于10℃。待温度稳定后将其转移至40℃水浴中反应5h,过滤,用5mL 三氟乙酸洗涤即得到1.360g白色固体,收率70.10%。
1H NMR(DMSO-d6,500MHz):11.16(s,1H,CH(NO2)2);13C NMR(DMSO-d6,125MHz):165.84(C5),131.41(CH(NO2)2);IR(KBr),v(cm-1):2 992,1 585,1 514,1 398,1 373,1 301,1 173,1 044,1 006,843,735;元素分析(C2HN7O6,%):理论值,C 10.97,H 0.46,N 44.76;实测值,C 10.93,H 0.43,N 44.65。
2 结果与讨论
2.1 ANT 的合成工艺改进
2.1.1 卤化盐对ANT 收率的影响
溶剂为DMF,温度为90℃,分别考察了在不添加卤化盐、加KBr、加KI 3种条件对反应时间、反应收率和产物纯度的影响,用薄层色谱(TLC)监测原料点消失所需要的时间,其中KBr和KI的摩尔数与氯丙酮相同。结果见表1。

表1 卤化盐对ANT 收率的影响Table 1 Effect of halide salt on the yield of ANT
表1显示,在未添加卤化盐的情况下,反应进行12h后,TLC观察原料点仍未消失;添加KBr或KI可以大大缩短反应时间。分析认为,在5-硝基四唑阴离子对卤代丙酮的亲核取代反应中,卤代丙酮的反应活性顺序为RI>RBr>RCl。当添加KI,产物外观颜色偏黄,其纯度比KBr稍低,可能是由于I-较强的还原性,使得部分KI和四唑环C5位上硝基发生了氧化还原反应所致。在反应中添加KBr,可获得较高的收率,且产物纯度较高。
2.1.2 溶剂对ANT 收率的影响
KBr作添加剂,考察了丙酮、乙腈、H2O 和DMF等溶剂对反应收率的影响,用丙酮和乙腈作溶剂时,反应温度分别为两者的回流温度,H2O 和DMF作溶剂时反应温度为90℃,结果见表2。

表2 溶剂对ANT 收率的影响Table 2 Effect of solvent on the yield of ANT
由表2可见,用水作溶剂时反应时间超过20h,原料点仍未消失;用丙酮和乙腈为溶剂时,所需反应时间较长,且收率较低;用DMF 作溶剂时,反应时间明显缩短。3种溶剂条件下,产率有所差异,纯度基本相当。因此,溶剂对反应速率有较大的影响,选DMF作溶剂较宜。
2.1.3 温度对ANT 收率的影响
DMF作溶剂,KBr作添加剂,反应温度分别为25、50、70和90℃,用TLC监测反应所需要的时间,结果见表3。

表3 反应温度对ANT 收率的影响Table 3 Effect of reaction temperature on the yield of ANT
表3显示,反应温度为25℃时,反应时间48h后,TLC观察原料点仍未消失;反应温度分别为50、70和90℃,原料均可在一定时间内反应完全,但比较而言,在90℃下反应20 min 即可完成。由此可见,反应速率随反应温度的升高而增大,结合产率和纯度,反应最佳温度为90℃。
综上所述,通过5-硝基四唑钠盐与氯丙酮反应制备ANT 的最佳条件为:反应添加的卤化盐为溴化钾,溶剂为DMF,反应温度为90 ℃。在此条件下,反应产率为74.02%,略低于文献报道值(75%)[3]。但与文献相比,本研究避免了使用毒性较大的溴丙酮,并大大缩短了反应时间。
2.2 ANT 的硝化-水解反应机理
通过羰甲基化合物的硝化-水解反应将偕二硝甲基引入唑环中,是偕二硝甲基唑类化合物合成的有效方法[6-8],ANT 分子中的亚甲基受到羰基和唑环的双重作用,具有较高的反应活性,易于被硝酰阳离子进攻,而亚甲基上的氢被硝基取代后,使得羰基易于水解离去,得到目标化合物HDNMNT。HDNMNT 经进一步硝化可得2-三硝甲基-5-硝基四唑(TNMNT),TNMNT 在-20℃以上迅速发生分解,要控制反应产物为偕二硝甲基唑类化合物HDNMNT,需控制硝化剂中的硝酰阳离子浓度和含水量。在ANT 的硝化-水解反应中,文献[3]使用浓硫酸、水和98%浓硝酸的混合体系,本研究直接使用65%的硝酸水溶液和浓硫酸作为硝化剂,简化了实验操作。
2.3 HDNMNT 的热分解行为
在升温速率10℃/min、氮气气氛中,用DSC 对HDNMNT 的热性能进行了分析,DSC曲线见图1。

图1 HDNMNT 的DSC曲线Fig.1 DSC curve of HDNMNT
由图1 可见,在0~350℃,HDNMNT 没有明显的吸热峰,表明在此温度范围内没有熔化过程,在温度119.85℃处出现一个尖锐的放热峰,判断其为分解峰,分解过程为放热过程,可见HDNMNT的热稳定性较差。
2.4 HDNMNT 及其阴离子DNMNT-的量子化学研究
运用量子化学方法,对HDNMNT 及其阴离子DNMNT-的几何构型和稳定性进行了理论研究,并计算了HDNMNT 的理论密度和固相标准生成焓,在此基础上,利用Kamlet-Jacobs公式[9]预估了其爆轰性能。
2.4.1 HDNMNT 和DNMNT-的几何构型
采用密度泛函理论(DFT)的B3LYP 方法[10-11],在6-31G**基组水平上对HDNMNT和DNMNT-的结构进行了全优化,经振动频率分析发现无虚频,表明优化结构为势能面上的极小点,为稳定构型。优化后的几何构型及原子编号见图2,键长、键角和二面角数据见表4。
由表4可见,四唑环上的N-N 和C-N 键长均处于典型的单键(0.148nm 和0.145nm)和双键(0.135nm 和0.125nm)之间,但HDNMNT 结构中C7-N8(0.1543nm)及C7-N9(0.1533nm)的键长则较一般单键长,表明其为最弱键。与DNMNT-的几何构型相比,HDNMNT 中C7上的H 原子被电子对取代后,导致C5-N6,N2-C7,C7-N8 和C7-N9 这4 个键之间的键长明显变短,同时所有硝基基团中的N-O 键均变长,表明偕二硝甲基中的两个硝基、碳原子(C7)和四唑环之间形成较大的共轭体系。对优化构型的自然键级轨道(NBO)分析所得的C、H、N 和O 之间的键级列于表4中,由表4可见,HDNMNT 的C7-N8的键级最小,而DNMNT-的C5-N6 的键级最小,键级最小的键易于断裂,为热解引发键。

图2 HDNMNT 和DNMNT-的几何构型Fig.2 The geometries of HDNMNT and DNMNT-

表4 HDNMNT 和DNMNT-的几何参数和键级Table 4 The optimized geometries and bond order of HDNMNT and DNMNT-
2.4.2 HDNMNT 和DNMNT-的C-N 键 离解能
采用密度泛函理论(DFT)的B3LYP 方法,在6-31G**基组水平上对HDNMNT 和DNMNT-结构中的C7-N8 和C5-N6 的键离解能进行计算(公式(1)),得到了硝基甲烷的C-N 的键离解能,结果见表5。由表5可见,HDNMNT 的C7-N8和C5-N6的键离解能分别为140.70kJ/mol 和266.47kJ/mol,DNMNT-的C7-N8和C5-N6的键离解能分别为255.73kJ/mol和294.94kJ/mol,通过比较可知,DNMNT-的热稳定性优于HDNMNT,且文献报道的HDNMNT 的铵盐和肼盐的热分解温度均大于HDNMNT[5],计算值与实验结果相吻合。

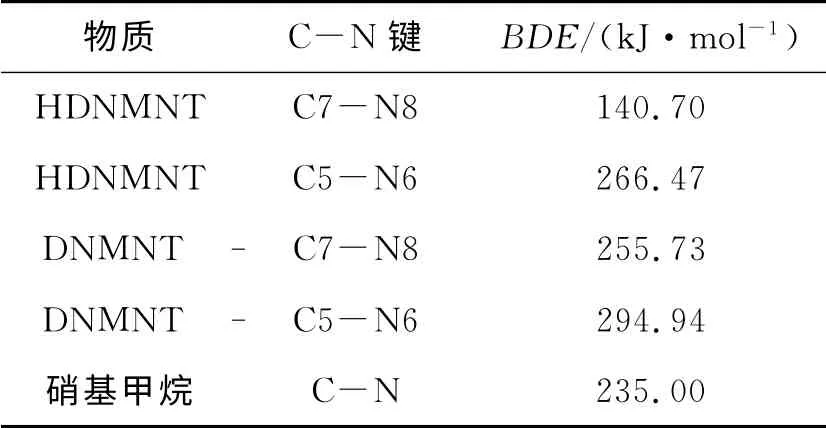
表5 硝基甲烷、HDNMNT 和DNMNT-中C-N 键的键离解能Table 5 BDEs of C-N bond in nitromethane,HDNMNT and DNMNT-
2.4.3 HDNMNT 的理论密度和固相标准生成焓
在B3LYP/6-31G**水平优化构型基础上,用Monte-Carlo法[12-13]计算分子体积,为了减小误差,取100次计算值的平均值为HDNMNT的摩尔体积,得Vm=114.55cm3,再根据Politzer等人[14]提出的公式求得HDNMNT的理论密度为1.880g/cm3。
使用原子化方案[15-17],利用完全基组方法(CBS-4M)[18-19]计算了298K 时HDNMNT的焓Ho(Molecule,298K),经振动分析确认无虚频,表明其均为稳定构型的焓值。298K 时,C、N、O原子的生成焓ΔfHo(Atoms,298K)取自文献值[20]。计算得到HDNMNT 的气相生成焓为389.89kJ/mol。利用Politzer等人[21]提出的公式和HDNMNT 的静电势参数计算了HDNMNT 的升华焓ΔHsub(116.85kJ/mol),进而求得HDNMNT 的固相生成焓ΔfHo(s,M,298K)为273.04kJ/mol。
2.4.4 HDNMNT 爆轰性能预估
对于组成为CaHbNcOd的含能材料,Kamlet-Jacbos公式是估算其爆轰性能较准确的经验公式。将HDNMNT 的理论密度和固相标准生成焓代入公式(2)和公式(3)中,计算结果见表6。并将其与RDX 的爆轰性能进行了比较。

式中:p为爆压,GPa;D为爆速,km/s;Q为每克含能材料的最大爆热,J/g;N为每克含能材料的气体爆轰产物物质的量,mol/g;为气体爆轰产物的平均摩尔质量,g/mol。
由表6可知,HDNMNT 的理论密度为1.88g/cm3,其生成焓为273.04kJ/mol,爆速为8 732m/s,爆压为35.37GPa。HDNMNT 的含氮量较高,正氧平衡,其爆轰性能与RDX 相当,是一种具有较高能量水平的含能化合物。

表6 HDNMNT 与RDX 的性能比较Table 6 Comparison of performance for HDNMNT and RDX
3 结 论
(1)以5-氨基四唑为原料,经过重氮化-取代反应、取代反应、硝化-水解反应三步合成得到2-偕二硝甲基-5-硝基四唑(HDNMNT),总收率39.95%。最佳合成ANT 的反应条件为:添加卤化盐为KBr,溶剂为DMF,温度为90℃。
(2)在0~350℃范围HDNMNT 没有熔化过程,仅存在一个放热分解峰,分解峰温为119.85℃,热稳定性较差。
(3)与HDNMNT 相比,DNMNT-的C-NO2中C-N 键的键离解能较高,热稳定性较好。
(4)HDNMNT 的理论密度为1.88g/cm3,固相标准生成焓为273.04kJ/mol,爆速为8 732m/s,爆压为35.37GPa,是一种具有较高能量水平的含能化合物。
[1]李冠琼,李玉川,马巧丽,等.富氮唑环类化合物的环加成合成研究进展[J].有机化学,2010,30(10):1431-1440.
LI Guan-qiong,LI Yu-chuan,MA Qiao-li,et al.Research progress in synthesis of nitrogen-rich zole-ring compounds by cycloaddition reaction[J].Chinese Jour-nal of Organic Chemistry,2010,30(10):1431-1440.
[2]刘晓建,张慧娟,林秋汉,等.唑类含能离子化合物的合成研究进展[J].火炸药学报,2010,33(1):6-10.
LIU Xiao-jian,ZHANG Hui-juan,LI Qiu-han,et al.Progress of study on the synthesis of zole energetic ionic compounds[J].Chinese Journal of Explosives and Propellants,2010,33(1):1431-1440.
[3]Semenov V V,Kanischev M I,Shevelev S A.Thermal ring-opening reaction of N-polynitromethyl tetrazoles:facile generation of nitrilimines and their reactivity[J].Tetrahedron,2009,65(17):3441-3445.
[4]Semenov V V,Shevelev S A,Mel'nikova L G.A general synthetic method for azolium and azinium dinitromethylides[J].Mendeleev Commun,1993,3(2):58-60.
[5]Semenov V V,Svyatoslav A S.Reactivity of the lownucleophilic N-dinitromethyl carbanion center in polynitromethylazoles[J].Mendeleev Commun,2010,20(6):332-334.
[6]Katritzky A R,Sommen G L,Gromova A V,et al.Synthetic routes towards tetrazolium and triazolium dinitromethylides[J].Chemistry of Heterocyclic Compounds,2005,41(1):111-118.
[7]Lim C H,Hong S,Chung K,et al.Synthesis and characterization of 5-dinitromethyltetrazole[J].Bull Korean Chem Soc,2008,29(7):1415-1417.
[8]Thottempudi V,Gao H,Shreeve J M.Trinitromethyl-substituted 5-nitro-or 3-azo-1,2,4-triazoles:synthesis,characterization,and energetic properties[J].J Am Chem Soc,2011,133(16):6464-6471.
[9]Kamlet M J,Jacobs S J.Chemistry of detonation I.a simple method for calculating detonation properties of CHNO explosives[J].Journal of Chemical Physics,1968,48(1):23-35.
[10]Becke A D.Density‐functional thermochemistry.III.The role of exact exchange[J].J Chem Phys,1993,98(7):5648-5653.
[11]Lee C,Yang W,Parr R G.Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density[J].Phys Rev B,1988,37(2):785-789.
[12]Qiu L,Xiao H M,Gong X D,et al.Crystal density predictions for nitramines based on quantum chemistry[J].J Hazard Mater,2007,141(1):280-288.
[13]Rice B M,Hare J J,Byrd E F C.Accurate predictions of crystal densities using quantum mechanical molecular volumes[J].J Phys Chem A,2007,111(42):10874-10879.
[14]Politzer P,Martinez J,Murray J S,et al.An electrostatic interaction correction for improved crystal density prediction[J]. Mol Phys,2009,107(19):2095-1101.
[15]Curtiss L A,Raghavachari K,Redfern P C,et al.Assessment of Gaussian-2and density functional theories for the computation of enthalpies of formation[J].J Chem Phys,1997,106(3):1063.
[16]Byrd E F C,Rice B M.Improved prediction of heats of formation of energetic materials using quantum chemical methods[J].J Phys Chem A,2006,110(3):1005-1013.
[17]Rice B M,Pai S V,Hare J.Predicting heats of formation of energetic materials using quantum chemical calculations[J].Comb Flame,1999,118(3):445-458.
[18]Ochterski J W,Petersson G A,Montgomery J A.A complete basis set model chemistry V.extension to six or more heavy atoms[J].J Chem Phys,1996,104(7):2598.
[19]Montgomery J A,Frisch M J,Ochterski J W,et al A complete basis set model chemistry VII.use of the minimum population localization method[J].J Chem Phys,2000,112(15):6532.
[20]Cox J D,Wagman D D,Medvedev V A.CODATA key values for thermodynamics[M].New York:Hemisphere Publishing Corp,1989.
[21]Politzer P,Murray J S,Grice M E,et al.Calculation of heats of sublimation and solid phase heats of formation[J].Mol Phys,1997,91(5):923-928.
[22]Wang R,Xu H,Guo Y,et al.Bis[3-(5-nitroimino-1,2,4-triazolate)]-based energetic salts:synthesis and promising properties of a new family of high-density insensitive materials[J].J Am Chem Soc,2010,132(34):11904-11905.
猜你喜欢
环境工程技术学报(2022年3期)2022-06-05
昆明医科大学学报(2021年4期)2021-07-23
化工管理(2021年7期)2021-05-13
化工管理(2021年10期)2021-04-25
分析化学(2019年3期)2019-03-30
安徽化工(2018年2期)2018-05-22
科学与财富(2016年7期)2016-03-25
绿色科技(2014年5期)2014-08-08
火炸药学报(2014年5期)2014-03-20
火炸药学报(2014年5期)2014-03-20
