
幽门螺杆菌及其细胞毒素相关蛋白A与胃癌形成的关系及可能机制
2013-01-17 12:21:24汪强武王启之于东红燕善军承泽农
中国组织化学与细胞化学杂志 2013年3期
汪强武 王启之* 于东红 燕善军 周 蕾 吴 炎 田 怡 承泽农
(1蚌埠医学院第一附属医院消化科,安徽233004;2蚌埠医学院病理学教研室,安徽233003;3广东省深圳市南山区中心医院消化科,深圳518052;4上海市闵行区中心医院消化科,上海201100)
胃癌是最常见的恶性肿瘤,探讨胃癌的病因学和发病机制一直是科研工作者研究热点。幽门螺杆菌(helicobacter pylori,Hp)与胃癌发生密切相关,Hp可通过诱发炎症、调节癌基因和抑癌基因的表达、诱导胃粘膜上皮增殖和凋亡异常及其代谢产物包括一些酶类、毒素和蛋白直接损害胃粘膜等形式引起胃癌发生。本文旨在检测胃癌、慢性浅表性胃炎和慢性萎缩性胃炎、肠上皮化生、不典型增生胃粘膜Hp及其CagA基因、P53、iNOS,探讨其与胃癌的相关性。
材料和方法
1.材料
标本:胃癌、慢性浅表性胃炎和慢性萎缩性胃炎、肠上皮化生、不典型增生组织取自我院2002年3月至2003年9月、2010年9月至2012年3月胃镜活检标本,胃癌377例,其中男262例,女115例;慢性浅表性胃炎306例,其中男232例,女74例;慢性萎缩性胃炎58例,其中男36例,女22例;肠上皮化生50例,其中男32例,女18例;不典型增生73例,其中男51例,女22例。所选病例胃镜下活检通过病理检查进一步证实。每例患者从病变处取六块活检标本,其中四块送病理检查,一块作尿素酶试验,一块装于无菌进口冻存管,冷冻于-80℃保存。快速尿素酶试验试剂盒:三明三强公司;幽门螺杆菌免疫印迹试剂盒:深圳伯劳特生物制品有限公司;P53、iNOS免疫组化试剂盒:北京中山实业有限公司;UNIQ柱式临床样品基因组抽提试剂盒:上海生工;CagA 基因引物1序列:5′-GAT AAC AGG CAA GCT TTT GAGG-3,引 物 2 序 列:5′-CTG CAA AAG ATT GTT TGC GAGA-3,由上海生工合成。
2.方法
2.1快速尿素酶试验
将钳取后的活检组织块迅速放入快速尿素酶试剂盒中,在30℃左右条件下观察试剂的颜色变化,如试纸由淡黄色变为红色,则为阳性;试纸不变色(仍为淡黄色)为阴性。胃粘膜组织Hp感染的轻、中、重度分别以+、++、+++表示。
2.2血清HpCagA抗体检测
每位患者于胃镜检查结束后抽取静脉血2ml,常温下分离血清,抗体检测按试剂盒中所述步骤进行,待阳性带显色清楚,干燥后对比标准带判断结果。
2.3病理切片染色找Hp
根据活检标本的病理号(见病历或活检标本病理报告出来后取胃镜报告单所登记)找到病理教研室的存档蜡块,并切病理薄片,行革兰氏染色,油镜下(×100)观察并计数,每例随机观察10-15个视野,分别取Hp平均数,Hp均数≥20个定为阳性,<20个或未查见Hp则为阴性,以排除因染色原因而造成的假阳性。Hp呈螺旋形,“S”形或海鸥形。
2.4HpCagA基因检测
取活检胃粘膜组织块(-80℃保存),未等解冻,用研钵研碎收集到1.5ml无菌离心管中,用200μlTE悬浮,用柱式临床样品基因组抽提试剂盒(操作步骤详见说明书)提取出DNA样品-4℃或-20℃保存。反应体系如下:10×PCR 缓冲液5μl,10μmol/L dNTP 1μl,引物1、2各取1.5μl,25mmol/L Mgcl2 3μl,TaqDNA酶1μl,Hp DNA提取液5μl,water(无核酸酶)32μl,石蜡油20μl覆盖。于94℃预变性5min后,94℃1min(变性)55℃1min(退火)72℃1.5 min(延长)35个循环后,72℃延伸10min。取PCR产物6μl于琼脂糖凝胶电泳。电泳条件87.5mv,30min于紫外线透射仪上观察,于349bp处出现橙黄色条带为CagA基因阳性(如图1所示)。
2.5免疫组织化学检查
采用SP法染色程序按SP法操作常规进行。对P53、iNOS抗原进行微波修复,同时在同一条件下以PBS代替一抗为阴性对照,以已知P53和iNOS蛋白阳性的胃癌为阳性对照。
3.统计学方法
采用χ2检验和四格表确切概率法,P<0.05为有统计学意义。
结 果
1.Hp和CagA基因的检测
革兰氏染色找Hp,结合快速尿素酶试验及免疫印迹试验检测血清HpCagA抗体三者有两项或以上阳性诊断为Hp感染。胃癌、慢性浅表性胃炎、慢性萎缩性胃炎、肠上皮化生、不典型增生标本中CagA基因PCR扩增产物检测结果见图1。
2.免疫组化SP法检测突变P53、iNOS结果判定
突变P53阳性为细胞核着棕黄色细颗粒(图2),iNOS阳性为细胞质着棕黄色细颗粒(图3),>10%的病变细胞为阳性时才判定为阳性。
3.不同胃疾病中Hp和CagA感染率比较
各种组织中Hp和CagA+株感染率的结果显示,胃癌组Hp感染率与浅表性胃炎、萎缩性胃炎和肠上皮化生组差异均无显著性(P>0.05),胃癌组Hp感染率与不典型增生组相比差异显著(P<0.05),而萎缩性胃炎、肠上皮化生和不典型增生组与浅表性胃炎组Hp感染率相比差异显著(P<0.05)。而不典型增生、胃癌组CagA+株感染率则高于浅表性胃炎组(P<0.01),肠上皮化生组CagA+株感染率则高于浅表性性胃炎组(P<0.05),胃癌组CagA+株感染率则高于萎缩性胃炎组(P<0.05),萎缩性胃炎组CagA+株感染率高于浅表性胃炎组,但统计学无差异(P>0.05)(表1)。
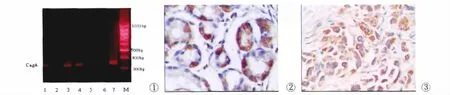
图1 胃癌、慢性浅表性胃炎、慢性萎缩性胃炎、肠上皮化生和不典型增生组织CagA基因PCR扩增产物。图2 胃癌细胞P53表达阳性(细胞核呈棕黄色细颗粒状)SP法×400图3 胃癌细胞iNOS表达阳性(细胞质呈棕黄色细颗粒状)SP法×400Fig.1The PCR amplification products of CagA gene in different stomach diseases.M:DNA分子量标准;1为慢性浅表性胃炎组织CagA基因阳性带;2、5为CagA基因阴性;3为慢性萎缩性胃炎组织CagA基因阳性带;4为肠上皮化生组织CagA基因阳性带;6为不典型增生组织CagA基因阳性带;7为胃癌组织CagA基因阳性带Fig.2The expression of p53in carcinoma cell of stomach by immunohistochemically staining(SP method×400)Fig.3The expression of iNOS in carcinoma cell of stomach by immunohistochemically staining(SP method ×400)
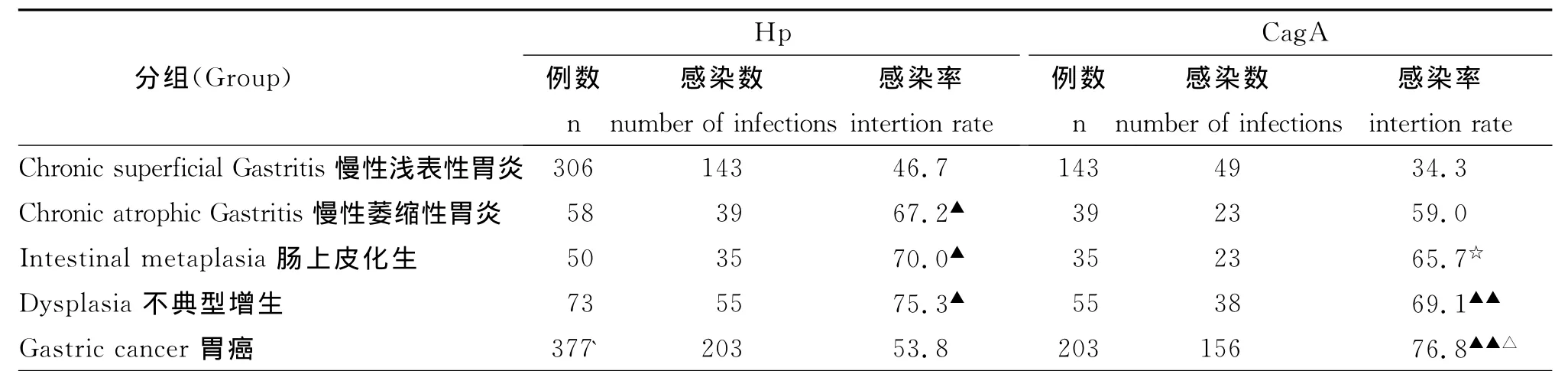
表1 不同胃疾病中Hp和CagA感染率比较Table 1 Comparisons of HP and CagA infection rates in different stomach diseases
4.不同胃疾病中CagA感染与突变P53、iNOS的表达关系
从慢性浅表性胃炎到胃癌演变过程中,CagA基因阳性组中的突变P53表达逐渐升高(34.3%-76.8%),CagA基因阳性组中的iNOS表达逐渐升高(75.5%-93.6%);除慢性浅表性胃炎外,慢性萎缩性胃炎、肠上皮化生、不典型增生和胃癌组CagA基因阳性组突变P53表达明显高于CagA基因阴性组突变P53表达(P<0.005-P<0.05);慢性浅表性胃炎、慢性萎缩性胃炎、肠上皮化生、不典型增生和胃癌组CagA基因阳性组iNOS表达均高于CagA基因阴性组iNOS表达(P<0.005-P<0.05)(表2)。

表2 不同胃疾病中CagA感染与突变P53、iNOS的表达关系Table 2 The relationship among CagA infection and expression of P53、iNOS in different stomach diseases
讨 论
幽门螺杆菌是一种螺旋状微需氧细菌,是慢性胃炎和消化性溃疡的主要致病因子,并且Hp感染与胃腺癌、胃粘膜相关性淋巴组织恶性淋巴瘤的发生密切相关[1]。
1990年,Cover等发现Hp培养液中存在相对分子量为12800的蛋白质,它虽不直接表达毒素活性,但与毒素活性表达密切相关,称为细胞毒素相关蛋白,即CagA。目前对Hp、CagA基因与胃十二指肠疾病的关系报道众说纷纭。
本研究发现浅表性胃炎Hp感染率46.7%,慢性萎缩性胃炎、肠上皮化生、不典型增生组织中Hp感染率逐渐升高。萎缩性胃炎、肠上皮化生、不典型增生是正常胃粘膜或浅表性胃炎向胃癌转化的关键病理形式,在它们的组织中检测到较高的Hp感染率,从而证明Hp与胃癌前病变及胃癌形成可能存在一定关系。但胃癌Hp感染率53.8%,与浅表性胃炎组Hp感染率相比无显著差异(P>0.05);可能是胃组织发生癌变后,胃内环境已不适合Hp的生存,致Hp死亡或迁移。
本研究还发现慢性浅表性胃炎CagA基因表达率为34.3%,从慢性萎缩性胃炎至胃癌CagA基因表达率逐渐升高。虽CagA本身无细胞毒活性,但与VacA的转录、折叠、运转功能等有关,CagA编码的细胞毒素蛋白可致宿主出现严重的炎症反应[2]。长期CagA+基因株感染使胃粘膜氧自由基、超氧化物生成增加,表面上皮细胞变性坏死、修复与增生、进而使腺体发生分化障碍与萎缩,形成肠上皮化生和不典型增生,最后可能形成胃癌[3]。CagA基因在胃癌前病变及胃癌高表达,机制可能为CagA+基因株产生多种毒素可导致胃粘膜细胞抑癌基因表达减弱和癌基因过度表达,导致基因突变而发生胃癌;国外学者Yamaoka等[4]进一步研究发现CagA基因3’端的超过3个的重复区域能加重组织损害使Hp更易在酸性环境下生存,与肠化生、萎缩及肿瘤有关。Williams等[5]报道CagA阳性Hp可能致胃炎、肠上皮化生、不典型增生和胃癌组织病理和染色体异常,通过 ROS (reactive oxygen species)介导肿瘤发生。各种胃疾病组织中CagA感染率逐渐升高,也说明了CagA基因参与各种胃癌前病变发生直至最后形成胃癌。
正常野生型p53基因的蛋白产物不稳定,半衰期仅20分钟,不能用免疫组化方法可以检测到。而突变型p53蛋白半衰期明显延长,用免疫组化方法能够检测到的p53蛋白均为突变型[6]。
关于Hp感染与P53突变报道不一[7]。Lima-VP等[8]研究发现胃癌形成机制有赖于Hp相关的凋亡参与和EB病毒参与的C-myc和Bax低表达,而P53突变只是胃癌形成中的一个独立事件。还有学者认为仅CagA阳性Hp感染在胃癌患者P53突变中起重要作用[9]。本研究结果表明突变P53表达在肠上皮化生和不典型增生组明显高于慢性浅表性胃炎,说明P53基因突变已经介入这种胃粘膜病变的形成,Hp阳性组和Hp阴性组相比,突变P53表达无显著差异,说明在胃粘膜病变的形成早期,P53基因突变可能与Hp感染无确定关系。本研究还显示突变P53在胃癌表达明显高于慢性浅表性胃炎,且胃癌CagA基因阳性中突变P53表达率高,说明CagA基因诱导P53基因突变并最终可能形成胃癌。Hp的CagA蛋白可通过影响胃粘膜细胞的增生和凋亡,增加DNA损伤的可能性,DNA的损伤又可引起突变P53的高表达,引起细胞增生和凋亡的失衡,进而形成胃癌[10]。
在慢性萎缩性胃炎、肠上皮化生、不典型增生和胃癌患者中,CagA基因阳性组较CagA基因阴性组的突变P53检出率高,CagA+株可能导致P53基因突变,细胞凋亡抑制,使慢性炎症导致的细胞增生无法控制,CagA+株感染与P53突变有非常显著的相关性,表明Hp的毒力越强,P53基因越易突变,胃癌发生率就越高[11]。新近 Kim N[12]运用多变量分析研究发现萎缩性胃炎的危险因素主要是Hp、年龄大于60岁、CagA基因和VacA基因感染及P53,也支持我们的观点。因此CagA基因在胃癌发生的起始阶段可能起了“启动子”作用,Hp诱导P53突变参与胃癌形成属于较晚期事件,且Hp致P53突变与CagA相关。
研究亦显示,在慢性浅表性胃炎、慢性萎缩性胃炎、肠上皮化生、不典型增生和胃癌组CagA基因阳性组iNOS检出率明显高于CagA基因阴性组iNOS检出率。
iNOS为一种诱生型酶,在大多数组织中不表达,但在一些炎性细胞因子、生长因子、内毒素作用下,呈高表达并发挥病理作用[13]。Hp感染可上调iNOS的表达,iNOS的表达与胃粘膜的炎症程度密切相关。Cho SO等[14]研究发现,在胃上皮细胞中,通过Ras介导,经AP-1活化,Hp可诱导COX-2和iNOS的表达,致使胃粘膜内NO含量增多。
Kaise等[15]发现,Hp感染在iNOS启动子长链CCTTT基因携带者中导致iNOS mRNA的高表达,故长链CCTTT基因携带者发生胃癌的危险性高。有学者认为萎缩性胃炎的腺体的消失是由于NO介导细胞凋亡所致,但损伤的细胞如逃避了凋亡机制,则可能发生癌变。
研究进一步表明,各种胃粘膜病变因CagA基因阳性引起iNOS升高,产生大量NO,NO参与胃癌的形成。NO可引起IL-8的产生;NO可直接损伤DNA;NO转化成硝酸盐和亚硝胺,对胃癌形成有一定作用;NO可通过VEGF促肿瘤血管形成,参与肿瘤的侵袭、生长、转移[16]。
HpCagA+株感染导致萎缩性胃炎、肠上皮化生、胃癌的过程中,首先激活了胃上皮细胞炎症细胞,产生大量活性氧、COX-2、iNOS、IL-6、IL-8等,这些炎症介质活化NF-KB;HpCagA的过度表达,增强胃粘膜细胞中iNOS和VEGF的表达,促进胃粘膜细胞癌基因c-erbB-2和ras的 活 化[17];HpCagA 的 表 达,致iNOS高表达,演变为肠化,最终形成肠型胃癌[18]。
赵亚刚等[19]研究发现,iNOS也可通过硝酸盐类、炎症介质、p53癌基因参与胃癌形成,增强胃癌的侵袭和转移力。
Perfetto B研究发现Th1类细胞因子如IFN-r表达iNOS基因,在P53、bax基因参与下,致Hp感染引起胃粘膜损害,同时避免宿主的防御反应[20]。Hp感染特别是CagA阳性患者胃粘膜iNOS增多和氧自由基释放,进而引起DNA损伤和P53基因突变。随着P53突变基因表达量的积累及其它致癌因素的共同作用,细胞便无限生长。进一步揭示了P53基因突变在胃癌形成中属晚期事件,当然亦不能忽视CagA基因、iNOS和氧自由基等因素在胃癌形成中作用。
胃癌的形成机制非常复杂,决不可能由一种因素引起或一个基因控制。Hp可能通过多种毒力因素及未发现的其它基因的综合作用促进突变P53过度表达致癌[21]。另一方面,Hp感染致胃癌在某些情况下缺乏生物学依据,且一部分胃癌组织CagA基因不表达,甚至Hp检测也为阴性,说明癌变机制有CagA基因外的其他机制参与,如P53、iNOS等基因。
[1]Cannizzaro R,De Paoli P.Helicobacter pylori eradication、endoscopic surveillance and gastric cancer.Am J Gastroenterol,2009,104(12):3100-3101
[2]Fereshteh J,Leil S.VacA genotypes of Helicobacter pylori in relation to cagA status and clinical outcome in Iranian populations Jpn.Infect Dis,2008,61:290-293
[3]叶超然.幽门螺杆菌及其CagA基因株感染与胃癌的相关性.广西医科大学学报,2002,19(2):204-205
[4]Yamaoka Y,El-Zimaity HM,Gutierrez O,et al.Relationship between the cagA 3’repeat region of Helicobacter pylori,gastric histology,and susceptibility to low pH.Gastroenterology,1999,117(2):342-349
[5]Williams L,Jenkins GJ,Doak SH,et a1.Fluorescence in situ Hybrid-isation analysis of chromosomal aberrations in gastric tissue:the potential involvement of Helicobacter pylori.Br J Cancer,2005,92(9):1759-1766
[6]范如英,李世荣,武子涛等.散发性大肠癌组织及粪便脱落细胞p53蛋白的检测.中国组织化学与细胞化学杂志,2001,10(4):443-445
[7]王启之,于东红,汪强武等.幽门螺杆菌相关胃疾病组织中p53、iNOS的表达.中国人兽共患病学报,2006,22(8):727-729,749
[8]Lima VP,de Lima MA,AndréAR,et al.H pylori(CagA)and Epstein-Barr virus infection in gastric carcinomas:correlation with p53mutation and c-Myc,Bcl-2and Bax expression.World J Gastroenterol,2008,Feb14;14(6):884-891
[9]Deguchi R,Takagi A,Kawata H,et al.Association between CagA Helico-bacter pylori infection and P53,bax,and transforming growth factor-beta-RⅡgene mutations in gastric cancer patients.Int J Cancer,2001,91(4):481-485
[10]杜雅菊,赵晶,关景明.胃癌组织中bcl-2和p53基因蛋白表达及其关系研究.中国现代医学杂志,2004,14(5):57-59
[11]童明宏,孙晨光.幽门螺杆菌感染、p53基因突变与胃癌的相关性研究.实用肿瘤杂志,2003,l8(5):393-395
[12]Kim N,Park YS,Cho SI,et al.Prevalence and risk factors of atrophic gas-tritis and intestinal metaplasia in a Korean population without significant gastroduodenal disease.Helicobacter,2008,Aug;13(4):245-255
[13]李荣,宋政军,王粉荣.iNOS、TGFβl、VEGF在胃癌中的表达及意义.现代肿瘤医学,2005,l3(5):609-611
[14]Cho SO,Lim JW,Kim KH.Involvement of Ras and AP-1in Helicobacter pylori induced expression of COX-2and iNOS in gastric epithelial AGS cells.Dig Dis Sci,2010,Apr;55(4):988-996
[15]Kaise M,Miwa J,Suzuki N,et al.Inducible nitric oxide synthase gene pro-moter polymorphism is associated with increased gastric mRNA expression of inducible nitric oxide synthase and increased risk of gastric carcinoma.Eur J Gastroenterol Hepatol,2007,19(2):139-145
[16]周小江,谢勇,吕农华.幽门螺杆菌感染及其相关疾病中COX-2、iNOS、NF-KB表达及其相互关系.陕西医学杂志,2007,36(3):289-292,295
[17]佟书娟,刘亚平,杨丹丹等.幽门螺杆菌在胃癌发生过程中的作用机制.第四军医大学学报,2005,26(24):2219-2222
[18]Rieder G,Hofmann JA,Hatz RA,et al.Up-regulation of inducible nitric oxide synthase in Helicobacter pylori-associated gastritis may represent an increased risk factor to develop gastric carcinoma of the intestinal type.Int J Med Microbiol,2003,Dec;293(6):403-412
[19]赵亚刚,张小瑞,吴炜景.诱导性一氧化氮合酶在幽门螺杆菌与胃癌关系中的研究进展[J].实用医学杂志,2009,25(7):1166-1167
[20]Perfetto B,Buommino E,Canozo N,et al.Interferongamma cooperates with Helicobacter pylori to induce iNOS-related apoptosis in AGS gastric adenocarcinoma cells.Res Microbiol,2004,May;155(4):259-266
[21]周丽雅,崔荣丽.幽门螺杆菌与胃癌.中华消化杂志,2010,30(7):497-499
猜你喜欢
西藏医药(2021年4期)2021-08-31 13:23:12
天津医科大学学报(2019年6期)2019-08-13 07:04:24
中国现代药物应用(2018年14期)2018-08-27 07:05:20
家庭用药(2018年11期)2018-01-23 12:29:52
中国民族医药杂志(2016年8期)2016-05-09 07:50:55
中国民族医药杂志(2016年8期)2016-05-09 07:50:48
中国民族医药杂志(2016年4期)2016-05-09 07:41:02
中国继续医学教育(2015年16期)2015-12-26 04:49:18
湖北科技学院学报(医学版)(2014年3期)2014-02-28 19:42:53
陕西医学杂志(2013年3期)2013-11-21 02:29:14
