
5-aza-dC对293A细胞增殖的影响
2013-01-12 05:35:04张颜波姜树原黄丽华孟宪梅
山东第一医科大学(山东省医学科学院)学报 2013年6期
刘 璐 张 伟 张颜波 姜树原 刘 友 黄丽华 孟宪梅 邵 国
(1. 包头医学院生物医学研究中心及基础医学部,内蒙古 包头 014060;2.包头医学院第二附属医院消化科,内蒙古 包头 014030; 3. 泰山医学院附属医院神经内科, 山东 泰安 271000)
抑癌基因启动子区的高甲基化在癌症发生发展过程中扮演了重要的角色,使DNA发生甲基化的酶是DNA甲基转移酶(DNA methyltransferase,DNMT),主要有三种:DNMT1、DNMT3A和DNMT3B。因此抑制DNMT活性的酶成为治疗癌症的潜在靶点。核苷酸类似物5-氮-2′脱氧胞苷(5-aza-2′deoxycytidine,5-aza-dC)可以参入复制的DNA双螺旋中而抑制DNMTs的活性,同时5-aza-dC也被认为可以参入RNA中抑制RNA的生物合成和代谢。5-aza-dC主要的抑癌作用表现为对细胞周期中S期的抑制作用[1]。本研究拟通过研究甲基转移酶抑制剂5-aza-dC对人胚肾细胞系293A的增殖的影响,为肾癌的治疗提供思路。
1 材料与方法
1.1 细胞
293A细胞购于上海细胞所。
1.2 试剂
RPMI1640 (GIBCO 公司), 5-aza-dC 和MTT (Sigma 公司), superscript III(Invitrogen 公司), 2 X real-time PCR Mix (南京凯基),其余试剂为国产试剂。
1.3 细胞培养及5-aza-dC处理
293A 细胞于含10% 新生牛血清的RPMI1640培养基, 37 ℃、5% CO2培养箱内培养。将5-aza-dC粉末溶于RPMI1640中配成浓度为1.0 mM的贮存液,过滤后-20℃保存。处理细胞时5-aza-dC的终浓度为1.0 μM。
1.4 MTT 法检测细胞增殖
调节细胞浓度每孔5×103接种入96 孔培养板, 贴壁后实验组加入1.0 μM 5-aza-dC后培养24 h开始检测,对照组加入等体积的PBS,连续检测4天,每天随机选3个孔加入20 μl MTT ( 5 mg/ml) 继续培养3 h, 去上清后加入150 μl DMSO 振荡10 min 后使用酶标仪检测, 以570 nm 波长处的光吸收值( A) 代表细胞活力,以培养天数(D)为横坐标,以吸光值为纵坐标,绘制细胞生长曲线。
1.5 流式细胞分析细胞周期
将细胞接种于96孔板中,当细胞生长到80%左右时,实验组用1.0 μM 5-aza-dC处理,对照组用等体积的PBS处理,72 h后PBS洗涤两次,然后细胞用0.5 ml的PBS悬浮,然后用 4.5 ml的70%乙醇固定过夜,离心收集细胞并用0.2 mg/ml 的propidium iodide (PI) 、0.1%的Triton X-100 和0.1 mg/ml RNase A室温避光处理30 min,用FACScan flow cytometer分析DNA含量,每个样本计数1×104细胞,每个样本3个复孔。细胞周期的分布用ModFit 3.0 程序分析。
1.6 5-aza-dC处理细胞的 DNMTs mRNA的 real-time PCR检测
Trizol提取处理72 h的实验组(1.0 μM 5-aza-dC)和对照组(等体积PBS) 细胞的总RNA, 按superscript III方法合成cDNA。根据Genebank序列,用Primer premier 5.0软件分别设计DNMT1,DNMT3A,DNMT3B和内参β-actin基因的引物。引物由上海生物公司合成,引物序列如下。DNMT1F:AACCTTCACCTAGCCCCAG;DNMT1R:CTCATCCGATTTGGCTCTTTCA;DNMT3A F:5-GACAAGAATGCCACCAAAGC;DNMT3A R:5-CGTCTCCGAACCACATGAC;DNMT3B F:AGGGAAGACTCGATCCTCGTC;DNMT3B R:GTGTGTAGCTTAGCAGACTGG;β-actin F:AGGTGAAGGTCGGAGTCA;β-actin R:GGTCATTGATGGCAACAA。Real-time PCR的条件与先前发表文章[2]相同:使用ABI的96孔板,每孔依次加入cDNA1 μl,2×mix 12.5 μl,3′和5′端引物(5 μmol/L)各1 μl,无菌水9.5 μl,在ABI 7900 real-time PCR反应仪上反应,每个样本设3个复孔。反应参数:94℃预变性3 min,再进行92℃,30 s;54.5℃,35 s;72℃,30 s(40 cycles),最后72℃延伸5 min停止反应。Real-time实验中DNMTs的CT值通过β-actin的CT值均一化,即ΔCT= CTDNMT s-CTβ-actin,而DNMTs mRNA相对丰度值以ΔΔCT值(DD value)表示,ΔΔCT=2-ΔCT。
1.7 数据分析及统计学处理
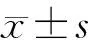
2 结 果
2.1 5-aza-dC处理对293A细胞增殖的作用
如图1所示,在接种于96孔板后4天时间内,5-aza-dC处理组和对照组细胞生长速度不同,第4天5-aza-dC处理组(0.0663±0.0034)是对照组(0.5453±0.0311)A值的12%左右(P<0.01,n=6)。结果提示,5-aza-dC处理组293A细胞的增殖能力明显降低。

图1 MTT分析293A细胞增殖
2.2 5-aza-dC处理对293A细胞周期的作用
5-aza-dC处理72 h后,细胞周期分布与对照组细胞的周期分布不同,大约有91%细胞停留在S期(图2),显示细胞阻滞与DNA合成期。


图2 流式细胞仪分析293A细胞周期
A:正常293A细胞;B:5-aza-dC处理的293A细胞
2.3 5-aza-dC处理对293A中DNMTs的mRNA表达的影响
如图3所示, 对照组293A细胞的DNMT1,DNMT3A和DNMT3B mRNA相对丰度分别为0.05594012±0.009,0.0362811±0.011和0.8751847±0.092。 1μM的5-aza-dC处理于293A细胞的DNMT1,DNMT3A和DNMT3B mRNA相对丰度分别为0.036810892±0.007,0.008603032±0.001和0.023717786±0.005。5-aza-dC处理使293细胞DNMTs mRNA的相对表达下降(P<0.05,P<0.01,n=3)。
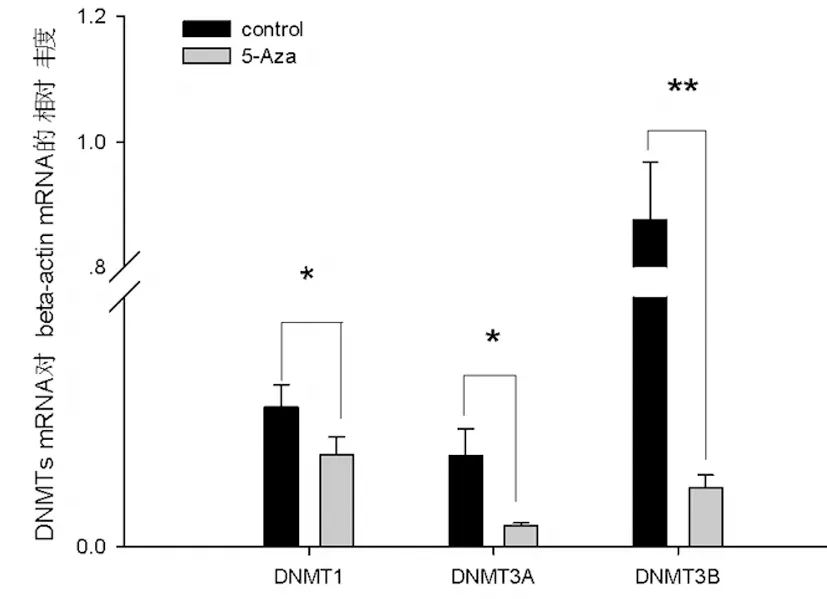
图3 real-time PCR分析5-aza处理293A细胞对DNMTs表达的影响
(*P<0.05 和**P<0.01,n=3)
3 讨 论
肾癌占据人类恶性肿瘤的2%~3%,与其他癌症相似,肾癌的发生伴随着一系列的遗传学事件和表观遗传学事件[3]。Avissar-Whiting等报道肾癌中LINE-1 和AluYb8这两个重复序列的甲基化程度与肿瘤的恶性程度成负相关,由于LINE-1 和AluYb8代表了基因组甲基化的情况,因此恶性程度高的肾癌的基因组的整体甲基化降低[4]。肾癌中抑癌基因的promoter区DNA甲基化变化情况是人们关注的一个热点,早在2003年Kawakami 和Morris两个研究小组的研究人员就报道了RASSF1A和P16等抑癌基因在肾癌生物样本和肾癌细胞系中甲基化的情况[5-6]。因此肾癌也表现出和其它癌症形式的甲基化特性:整体甲基化水平降低和抑癌基因启动子区甲基化水平升高。5-aza-dC是一种核苷酸类似物,在DNA复制过程中参入DNA与DNMTs共价结合形成复合物从而抑制DNMTs的甲基转移活性。5-aza-dC 可使多种因甲基化而失活的抑癌基因重新表达,并且可以诱导凋亡,体外实验证明其可抑制多种肿瘤细胞的生长[1]。所以,5-aza-dC对肾脏肿瘤细胞的作用,特别是DNMTs的影响对明确其抑癌机理有重要作用。
5-aza-dC对DNMT1有抑制作用非常明确[7-8],但对DNMT3A和DNMT3B的作用报道比较少,5-aza-dC处理大肠癌HCT116细胞系会降低DNMT1和DNMT3A的mRNA的表达,但对DNMT3B没有影响[9]。而5-aza-dC处理子宫内膜癌细胞系Ishikawa,导致DNMT3B的表达降低[10]。可见不同的细胞系中DNMTs对5-aza-dC反应不同。在我们的研究中用5-aza-dC处理胚肾细胞系293A发现DNMT1、DNMT3A和DNMT3B mRNA都降低,处理后的DNMT1、DNMT3A和DNMT3B mRNA的丰度分别约是处理前的2/3、1/4和1/37,5-aza-dC处理胚肾细胞系293A使DNMT3B降低的最多。
5-aza-dC处理293A细胞72 h后,细胞周期与对照相比,出现了明显的差异,大约90%的细胞停留在G2/S期,由于细胞周期的停滞,导致细胞增殖受到抑制。而在293A细胞中5-aza-dC处理使DNMT3B大幅度的下降,同时DNMT3A下降了75%,DNMT1、DNMT3A和DNMT3B的变化可能是293A细胞周期阻滞的原因。Oka等报道了胚胎干细胞对5-aza-dC的反应,DNMT3A和DNMT3B双缺失的胚胎干细胞对5-aza-dC抵抗能力比野生型、DNMT1缺失、DNMT3A缺失和DNMT3B缺失的干细胞要高。当外源性的DNMT3A或DNMT3B基因转移到DNMT3A和DNMT3B双缺失的胚胎干细胞中后,其对5-aza-dC敏感性恢复[11]。因此在293A细胞中,DNA-DNMT3A或DNA-DNMT3B在5-aza-dC诱导的细胞反应中可能扮演了重要角色。
在本研究中,我们只观察了5-aza-dC 处理对293A细胞DNMT1、DNMT3A和DNMT3B mRNA表达的影响和细胞周期的影响,为了明确DNMT3A和DNMT3B的变化在对293A细胞周期中的作用,需要我们做大量的工作进一步去明确。
[1] Yang Q, Shan L, Yoshimura G, et al. 5-aza-2'-deoxycytidine induces retinoic acid receptor beta 2 demethylation, cell cycle arrest and growth inhibition in breast carcinoma cells[J]. Anticancer Research, 2002, 22 (5):2753-2756.
[2] 姜树原, 张颜波, 隋欣,等. 宫颈癌组织中DNA甲基转移酶1、3A和3B mRNA的表达[J]. 泰山医学院学报, 2011, 8 (5):321-323.
[3] Vidaurreta M, Maestro ML, Sanz-Casla MT, et al. Inactivation of p16 by CpG hypermethylation in renal cell carcinoma[J]. Urologic Oncology, 2008, 26 (3):239-245.
[4] Avissar-Whiting M, Koestler DC, Houseman EA, et al. Polycomb group genes are targets of aberrant DNA methylation in renal cell carcinoma[J]. Epigenetics, 2011, 6 (6):703-709.
[5] Kawakami T, Okamoto K, Ogawa O, et al. Multipoint methylation and expression analysis of tumor suppressor genes in human renal cancer cells[J]. Urology, 2003, 61 (1):226-230.
[6] Morris MR, Hesson LB, Wagner KJ, et al. Multigene methylation analysis of Wilms' tumour and adult renal cell carcinoma[J]. Oncogene, 2003, 22 (43):6794-6801.
[7] Patel K, Dickson J, Din S, et al. Targeting of 5-aza-2'-deoxycytidine residues by chromatin-associated DNMT1 induces proteasomal degradation of the free enzyme [J]. Nucleic Acids Research, 2010, 38 (13):4313-4324.
[8] Maslov AY, Lee M, Gundry M, et al. 5-Aza-2'-deoxycytidine-induced genome rearrangements are mediated by DNMT1[J]. Oncogene, 2012, 31 (50):5172-5179.
[9] Schneider-Stock R, Diab-Assef M, Rohrbeck A, et al. 5-Aza-cytidine is a potent inhibitor of DNA methyltransferase 3a and induces apoptosis in HCT-116 colon cancer cells via Gadd45-and p53-dependent mechanisms[J]. The Journal of Pharmacology and Experimental Therapeutics, 2005, 312 (2):525-536.
[10] Cui M, Wen Z, Chen J, et al. 5-Aza-2'-deoxycytidine is a potent inhibitor of DNA methyltransferase 3B and induces apoptosis in human endometrial cancer cell lines with the up-regulation of hMLH1[J]. Medical Oncology (Northwood, London, England), 2010, 27 (2):278-285.
[11] Oka M, Meacham AM, Hamazaki T, et al. De novo DNA methyltransferases Dnmt3a and Dnmt3b primarily mediate the cytotoxic effect of 5-aza-2'-deoxycytidine[J]. Oncogene, 2005, 24 (19):3091-3099.
猜你喜欢
海南医学(2016年8期)2016-06-08 05:43:00
健康管理(2016年2期)2016-05-30 21:36:03
山东医药(2015年14期)2016-01-12 00:39:43
中国医疗美容(2015年1期)2015-07-12 10:06:52
医学研究杂志(2015年9期)2015-07-01 17:27:46
医学研究杂志(2015年7期)2015-06-22 11:00:39
天津医科大学学报(2015年3期)2015-06-05 12:21:49
江苏大学学报(医学版)(2015年2期)2015-04-17 06:49:51
中国当代医药(2015年9期)2015-03-01 02:02:01
中国医药导报(2015年26期)2015-02-28 22:07:44
