
橡胶增韧塑料机理研究进展
2012-12-23 09:04王荣伟
合成树脂及塑料 2012年2期
王荣伟
(中国石油化工股份有限公司上海石油化工研究院,上海市 201208)
橡胶增韧塑料机理研究进展
王荣伟
(中国石油化工股份有限公司上海石油化工研究院,上海市 201208)
介绍了橡胶增韧塑料机理演进及其与不同链结构基体树脂断裂行为的关系,描述了银纹和裂缝区别,详细地介绍了不同基体树脂的分子链参数与橡胶分散相形态参数的定量关系,同时强调基体树脂的链参数既控制聚合物/橡胶共混物的本征韧性,又决定了橡胶分散相的适宜形态。
塑料 橡胶 增韧 塑性 脆性 银纹 裂缝 屈服
1 理论的演进
虽然用天然橡胶改性聚苯乙烯(PS)的本体和溶液沉淀技术雏形于1925年就已形成[1],但较为令人信服的、定性的橡胶增韧作用理论解释却出现在1950—1960年。之前,橡胶以力学阻尼方式吸收冲击能的观点较为人们所接受。该观点的缺陷在于不能解释应力发白和应变大的形变。
1.1 微裂缝理论
1956年,Merz等针对高抗冲聚苯乙烯(HIPS)(用丁苯橡胶制备)被拉伸时横截面基本无收缩和密度下降8%的情况,发现材料内部产生了空穴,认为拉伸时材料出现应力发白现象是材料内部产生的空穴对光散射引起的。另外,他们提出了应变时材料内部产生许多很细微裂缝的概念以及第一种增韧理论(微裂缝理论),即部分橡胶颗粒横跨裂缝之上,阻止了裂缝迅速发展;形变过程中,橡胶颗粒消耗了能量,从而提高了材料的韧性。据此,假设橡胶颗粒处于裂缝增长方向的正面,那么,被吸收的冲击能等于基体树脂断裂能和橡胶颗粒破碎所需做功的总和。Newman等[2]估算后认为,橡胶颗粒在拉伸中吸收的能量很少,约占总吸收能的1/10。显然,基体树脂的作用在微裂缝理论中未被充分重视。该理论不足之处还在于不能解释增韧聚氯乙烯(PVC)的断裂行为。
1.2 多重银纹化理论
图1是聚2-乙烯基吡啶(PVP)/PS合金的裂缝和银纹透射电子显微镜照片。

图1 PVP/PS合金的裂缝、银纹、界面透射电子显微镜照片[3]Fig.1 Transmission electron micrographs of the crack,craze and interface in PVP/PS alloy
早在1949年,Sauer等研究银纹和裂缝后,提出了银纹在整个试样截面的扩展使PS能够承受一定负荷的观点。X射线衍射证据使他们得出了银纹由取向聚合物和散布其间的空穴组成的结论。1961年,根据去除负荷后试样银纹的X射线衍射特征未变现象,Bessonov等得出了相同结论。1960年,Schmitt等将橡胶颗粒视作应力集中点,认为这些点会产生银纹,银纹增长需消耗较多能量,同时,银纹应力场相互干扰而减弱了银纹增长的前沿应力,从而导致银纹终止。由此他们还认为,应力发白是银纹引起而不是裂缝所致。1962年,Spurr等[4]用光学显微镜和扫描电子显微镜观察到银纹中含有取向的聚合物,提出了银纹诱发、增长、终止三步骤的应力导致银纹形成的机理,并指出通过加热或溶剂熏蒸方式可以消除银纹。同年,Kambour[5]进一步指出,玻璃态聚合物形变过程中,银纹化先于断裂而发生。他也观测到了银纹愈合现象,实验计算得出聚碳酸酯(PC)的银纹中含有50%~60%聚合物。

银纹是材料受力后发生非均匀塑性形变的一种产物。银纹具有的特点[6]包括:1)长×宽可达100 μm×10 μm;2)其长轴与应力方向垂直;3)它们既不是裂缝也不是空穴。它们内部含非晶形、取向的微纤,直径为5~50 nm[7-8],两端与基体相连,微纤与应力平行,与银纹长轴垂直;4)通过加热或溶剂熏蒸方式可以消除银纹。
由于基体、微纤、空气折射率不同和银纹本身的尺度大于可见光的波长(0.4~1.1 μm),因此,银纹能产生光散射并导致试样应力发白现象[6]。
在微裂缝理论和以上对银纹理解的基础上,Bucknall等[9]于1965年提出了多重银纹化理论。该理论进一步地阐明,橡胶颗粒既可以引发银纹又可以控制银纹的增长。在应力作用下,银纹发生于最大主应力点,主应力点集中于橡胶颗粒的赤道区域;银纹沿最大主应力平面增长。当银纹尖端的应力降低至银纹增长所需的临界水平以下时、或遇到一个大的橡胶颗粒或其他障碍物时,银纹增长被终止[8-9]。
多重银纹化理论能成功地解释PS和HIPS的抗冲击和拉伸性能[10],包括应力发白、密度下降和无侧面收缩的伸长。橡胶含量和颗粒尺寸、界面黏结力和温度等因素对材料性能的影响都可借鉴该理论来说明。但是,它不能解释丙烯腈(AN)-苯乙烯-丁二烯三元共聚物(ABS)和增韧PVC拉伸屈服后出现的细颈化现象,也不能解释以PVC为基体的材料中细颈化伸长后无应力发白现象。
1.3 屈服膨胀理论(剪切屈服理论)
Newman等[2]在研究ABS和PVC拉伸形变时提出了屈服膨胀理论。他们认为,在橡胶颗粒周围基体树脂相中存在的流体静拉伸应力促使基体树脂相的自由体积增大,从而降低了玻璃化转变温度(Tg),使它能发生塑性流动。形成这种应力的原因有:1)热收缩差。橡胶热膨胀温度系数比基体树脂大,材料成型后由高温冷却至室温时,橡胶颗粒收缩比基体树脂大,故形成这种应力;2)力学效应。橡胶泊松比大,横向收缩大;基体树脂泊松比较小,横向收缩较小,故可形成这种应力。显然,上述原因分析的前提是两相界面黏结力要强。
根据屈服膨胀理论以及ABS和苯乙烯-AN共聚物(SAN)的屈服强度等参数,Bragaw推导并计算发现该理论所给出的体积膨胀只相当于升温12℃所产生的体积膨胀。而在室温下,要使Tg约90℃的SAN出现屈服,那么该理论所预计的体积膨胀太小了。再者,如果用刚性颗粒或者空气泡替代橡胶颗粒,则该理论预计的结果是,橡胶颗粒增韧效果不及前两者。另外,该理论不能解释橡胶颗粒增韧基体树脂时出现的应力发白、密度变化、无明显细颈化伸长等现象。
1.4 剪切屈服-银纹化理论(剪切带-银纹化理论)
剪切屈服-银纹化理论以多重银纹化理论和剪切屈服理论为基础。在材料形变过程中,银纹化和剪切屈服是并存的两种能量耗散方式。材料和条件不同,两种方式的贡献不同。HIPS在常温下受力形变以银纹化耗散能量为主,剪切屈服的贡献较少;在高温下会出现细颈化现象。ABS在常温下受力形变时银纹化和剪切屈服同时出现,所以,既出现应力发白又出现细颈化现象。
剪切屈服-银纹化理论不仅考虑了材料形变时的两种能量耗散途径,而且银纹还具有相互终止、诱发和终止剪切带功能,剪切带具有终止银纹的功能(见图2)。所以,该理论既考虑了橡胶颗粒的作用,又注意到了基体树脂的影响。
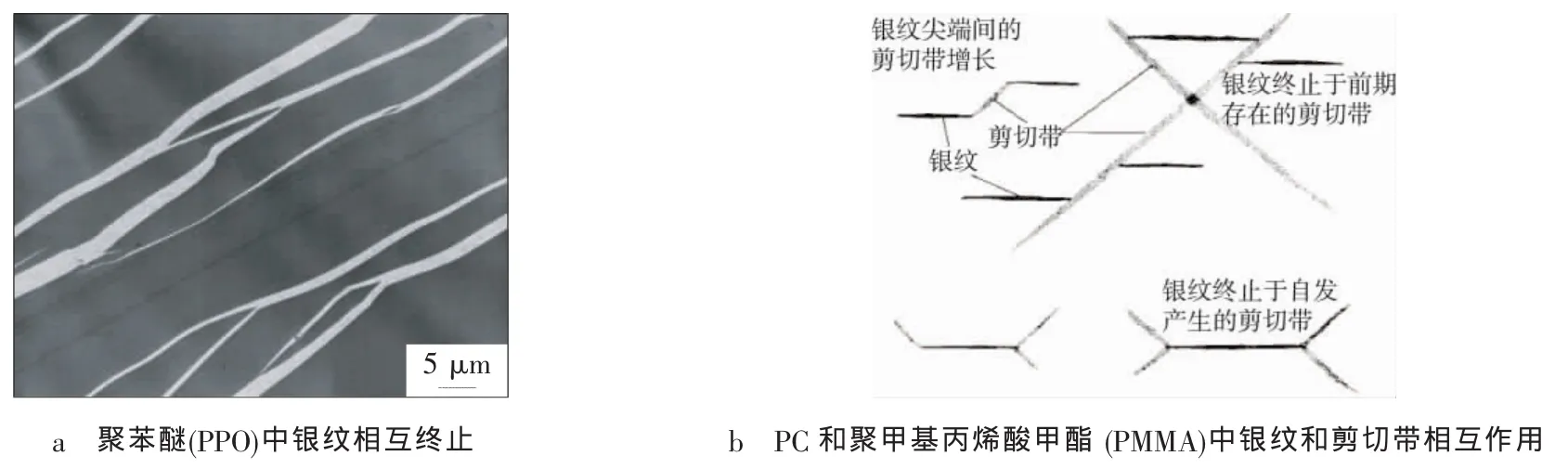
图2 银纹的相互终止、诱发和终止剪切带[11]Fig.2 Mutually terminated crazes in poly(2,6-diphenyl-l,4-phenylene oxide),interactions of crazes and shear bands in PMMA and polycarbonate
2 增韧机理的定量描述
Wu Souheng[12]指出,聚合物及其共混物的脆性/塑性(银纹/屈服)行为与外在因素(速率、温度、应力状态、缺口和试样几何形状)和内在因素(相形貌和链结构)有关,依据变形时能量耗散或破损可通过银纹化、屈服或两者结合方式来实现。Wellinghoff等[13]将玻璃态聚合物分为倾向于银纹化的乙烯基聚合物和倾向于屈服的主链含芳基聚合物。Wu Souheng[14]直接将聚合物分为脆性和准塑性。脆性聚合物以银纹化为破损方式,其裂缝引发能量低(即无缺口韧度低)以及裂缝增长能量也低。准塑性聚合物以屈服为破损方式,其裂缝引发能量高(即无缺口韧度高)而裂缝增长能量低(即缺口韧度低)。兼有银纹和屈服特征的聚合物破损方式介于上述两者之间。银纹和屈服构成了橡胶增韧玻璃态聚合物(基体树脂)机理的两个要素[15]。
为揭示相形态、链结构与增韧的关系,Wu Souheng定义了两个微观结构参数作为基体树脂的本征变量:一个是链缠结密度(ve)[见式(1)];另一个是链特征比(C∞)[12][见式(2)]。
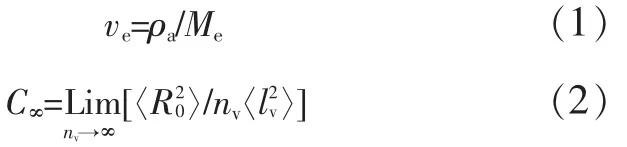
两者的关系见式(3)。

式中:ρa是非晶形聚合物密度;Me是分子链上两邻近缠结点间的相对分子质量;〈〉是无扰链均方末端距;〈l2v〉是统计链节均方长度;nv是一个分子链上统计链节数,Mv是统计链段的相对分子质量。
从图3看出:纯基体树脂的银纹应力(σz)与ve1/2成正比。
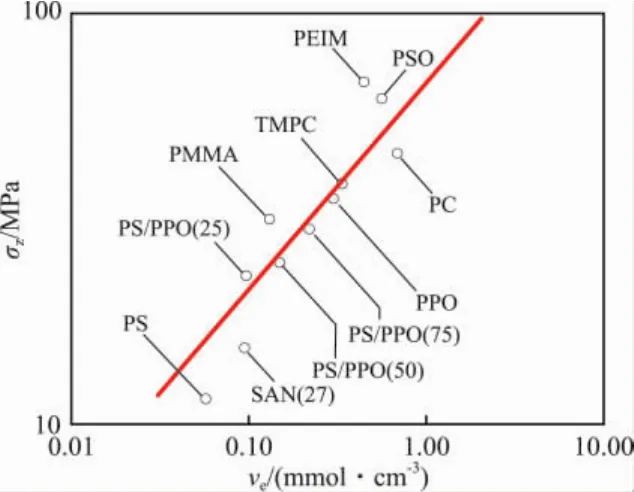
图3 纯基体树脂的σz~veFig.3 Crazing stress σzversus entanglement density vefor virgin polymer glasses
从图4看出:纯基体树脂的归一化屈服应力{σy}等于σy/[δ2(Tg-T)](σy是屈服应力,δ2是内聚能密度,T是测试温度),与C∞成正比。
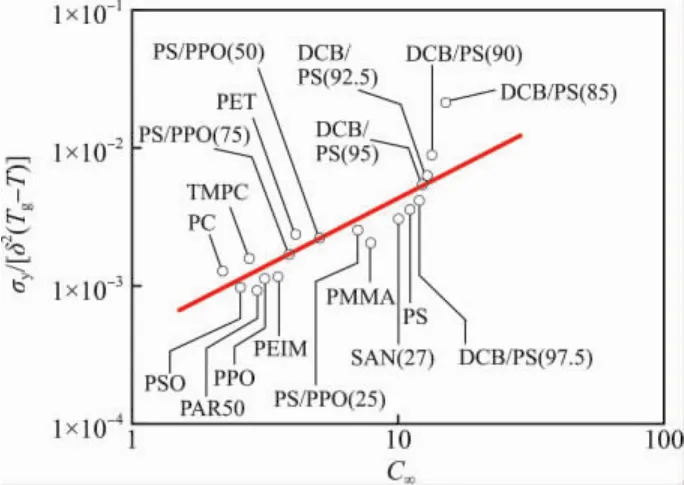
图4 纯基体树脂的σy/[δ2(Tg-T)]与C∞关系,Fig.4 Normalized yield stress σy/[δ2(Tg-T)]versus characteristic ratio C∞for virgin polymer glasses
基于上述关系,Wu Souheng给出了控制银纹-屈服行为的分子判据[见式(4)]。
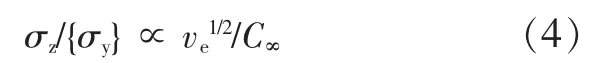
根据式(3),将式(4)整理为式(5)和式(6)。
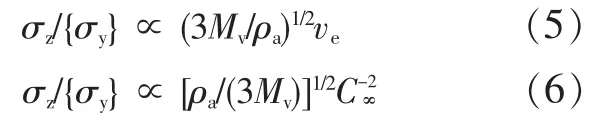
式(5)和式(6)中,对许多聚合物而言,(3Mv/ ρa)1/2是常数。因此,σz/{σy}可表示成与ve或C∞有关。上式表明,银纹与屈服机理间的竞争决定了聚合物的脆性/塑性行为。当σz低于屈服应力,聚合物倾向银纹化;当屈服应力低于σz,聚合物倾向于屈服。当聚合物ve低且C∞值大时,σz/{σy}比值低,聚合物倾向银纹化;反之,当聚合物ve大且C∞值低时,σz/{σy}比值高,聚合物则倾向于屈服。
表1、表2分别列出了一些脆性和准塑性聚合物的C∞和ve。表1以ve值增加方式排序;表2以C∞值递减方式排序。聚合物的本征塑性随C∞降低而增加。对无规行走链而言,C∞=1;对四面体价键构成的自由旋转链,C∞=2。
综上所述,Wu Souheng认为,基体聚合物(基体树脂)的链参数既控制聚合物/橡胶共混物的本征韧性,又决定橡胶颗粒的适宜形态。为此,Wu Souheng借ve和C∞的不同组合,将聚合物共混物分为三类。
2.1 脆性基体树脂构成的共混物
ve<0.15 mmol/cm3,C∞>7.5。典型的聚合物有PS,SAN25[w(AN)为25%],PMMA。从本征脆性而言,PS最大,SAN25次之,PMMA最小。三种纯聚合物的形变都受银纹化开裂机理控制。然而,共混物中,分散的橡胶颗粒能促进和/或改变基体树脂的开裂机理。分散的橡胶颗粒在PS中诱发多重银纹,在SAN25中同时诱发银纹和屈服,在PMMA中开裂机理却由无橡胶时银纹化变成剪切屈服。添加同体积的橡胶颗粒,增韧SAN的冲击强度最高。因此,ve≅0.1 mmol/cm3的脆性聚合物可以制成超韧共混物。
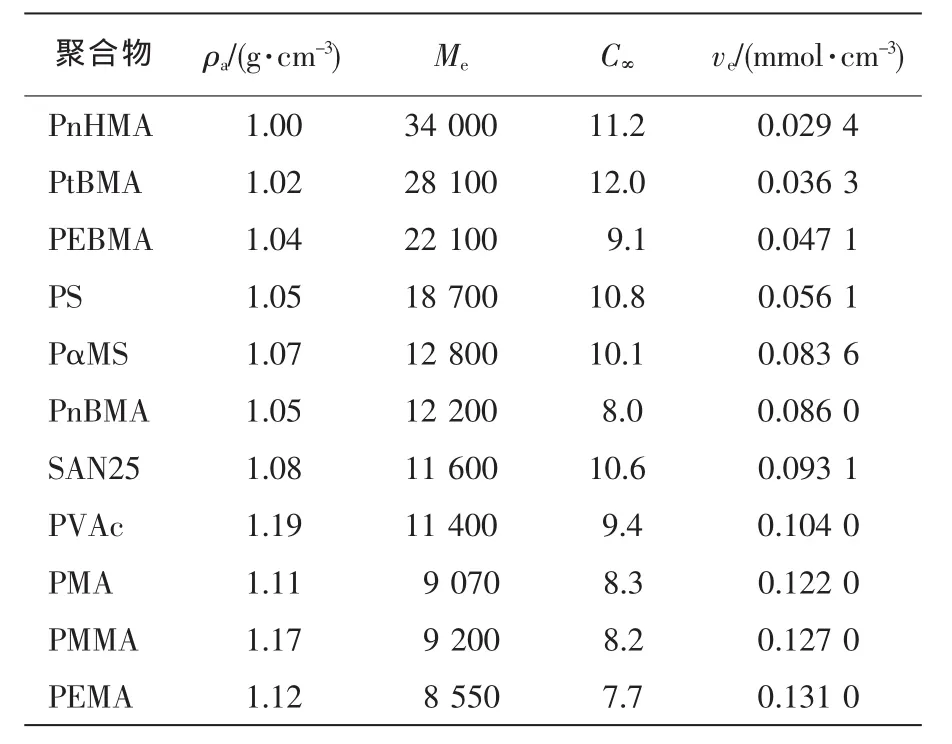
表1 一些非晶形脆性聚合物的链参数[12,14]Tab.1Chain parameters for some brittle polymers in amorphous state
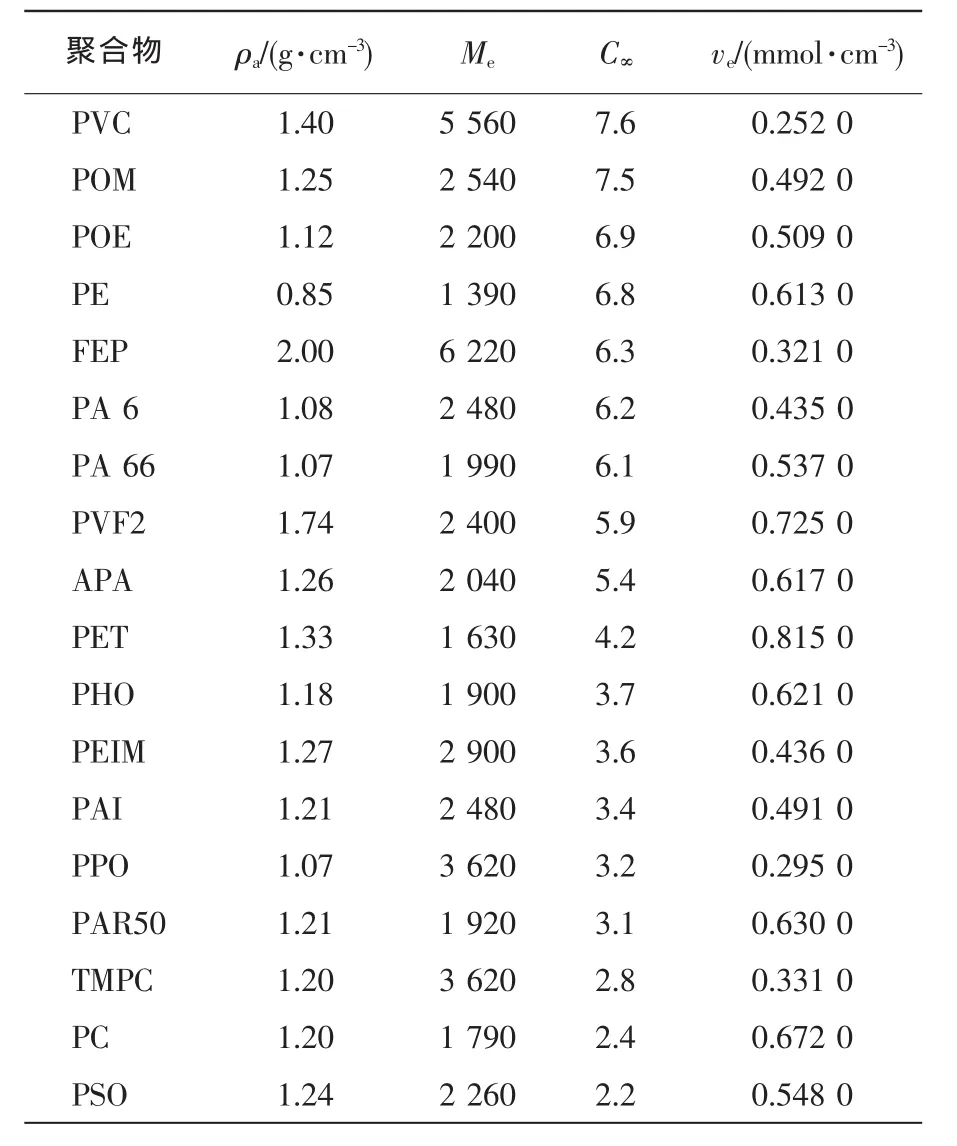
表2 一些非晶形准塑性聚合物的链参数[12,14]Tab.2Chain parameters for some ductile polymers in amorphous state
Wu Souheng根据Keskkula等[16]用甲基丙烯酸甲酯-丁二烯-苯乙烯树脂增韧SAN的实验结果(见图5)确定了ve与SAN组成的关系[见式(7)]。
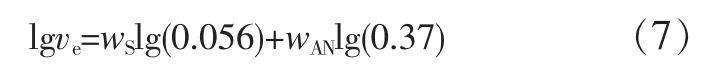
式中:wS表示SAN中苯乙烯质量分数;wAN表示SAN中AN质量分数。
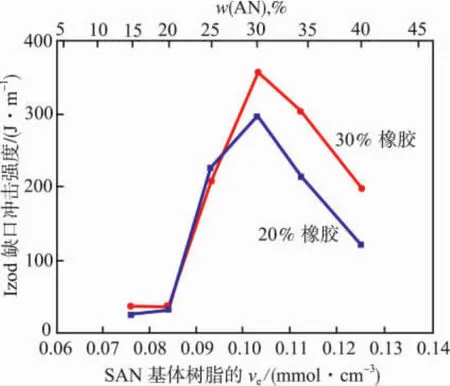
图5 韧性与SAN的ve和w(AN)关系Fig.5 Toughness versus veand mass content of acrylonitrile for SAN/rubber blends
图6表明小颗粒的橡胶分散相能有效地引发屈服,大颗粒有利于引发银纹。针对此三种聚合物共混,Wu Souheng用数学方法将最佳橡胶粒径(d0)与ve进行了关联[见式(8)]。
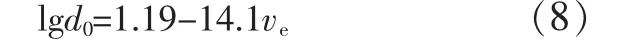
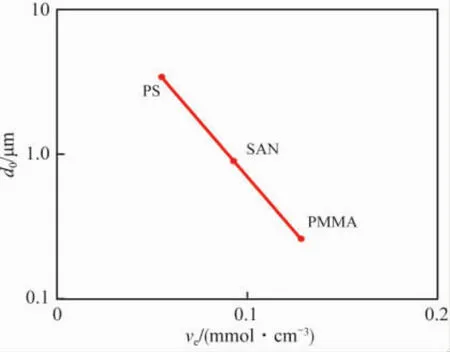
图6 基体树脂ve与d0的关系[17]Fig.6 Optimum rubber particle size for the toughening of brittle matrices versus the entanglement density of matrices
2.2 准塑性基体树脂构成的共混物
ve>0.15 mmol/cm3,C∞<7.5。典型的聚合物有PA 66,PET,PC。从本征塑性而言,PC最大,PET次之,PA 66最小。所有准塑性纯聚合物及其共混物的形变都受屈服开裂机理控制。在研究增韧PA时,Wu Souheng发现临界橡胶颗粒大小(dc)随橡胶含量而变,另外,还研究发现,临界基体树脂韧带厚度(τc)与橡胶体积分数无关,但与应力状态、速率、温度有关。由此,他给出了dc和τc的关系[见式(9)]。
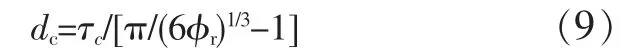
式中:φr是橡胶体积分数。图7表明,τc与基体树脂的C∞相关,以最小二乘法线性回归得式(10)。
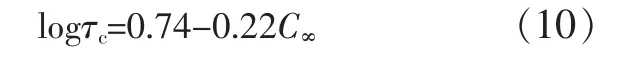
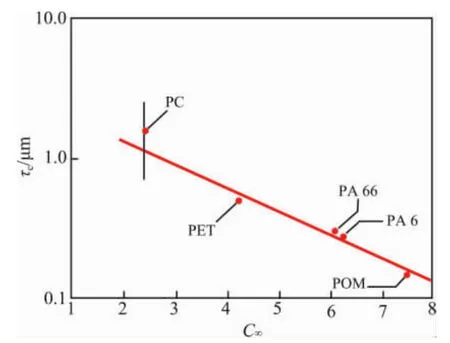
图7 准塑性基体树脂脆-韧转变点的τc~C∞Fig.7 Criticalmatrixligamentthicknessτc(criticalsurface-to-surface interparticle distance)for the onset of brittle-tough transition in pseudoductile matrices versus characteristic ratio C∞of the matrix
针对上述准塑性基体树脂存在τc的现象,Wu Souheng借逾渗理论(即形态连通度概念)成功地解释了脆-韧转变。逾渗理论的要点是:当系统的成分或某种意义上的密度变化达到一临界值(称为逾渗阈值)时,系统的相互关联性会出现陡然的变化,且满足一定的标度或相似关系[18]。增韧机理与橡胶颗粒的空穴化有关,这种空穴化可以释放膨胀应力,因而使得薄基体树脂韧带(τ<τc)(τ为基体树脂韧带厚度)易于发生局部屈服。当薄基体韧带相互连通而形成弥散网络时,屈服过程易于扩展且可弥散于整个变形区域。当发生这种现象时,共混物将呈韧性。Wu Souheng用“应力体积”逾渗模型对PA/橡胶共混物脆-韧转变进行了计算,证明了该系统符合两个标度关系:应力体积的阈值是常数;在临近脆-韧转变点,材料韧性服从近阈值临界标度关系。Wu Souheng还指出,逾渗模型很好地预期了增韧准塑性基体树脂时用单分散橡胶颗粒比多分散的有效;不对称(大的长宽比)橡胶颗粒(如棒状、带状、小板状、蛛丝状和网状)比球状更有效;形成孤立的橡胶颗粒集群絮结物是有害的,但部分成相互连通的网状絮结物是有益的,因为其降低了应力体积的阈值。
针对Wu Souheng提出的τc概念,Bucknall等[19]讨论了PA/橡胶体系的增韧机理,建立了与橡胶颗粒大小有关的模型,指出Wu Souheng提出的概念不合理的理由。但他又指出,文章中设立的模型未考虑橡胶含量变化、温度、应变速率等因素。
2.3 介于脆性和准塑性的基体树脂构成的共混物
ve≅0.15 mmol/cm3,C∞≅7.5。典型的聚合物有PMMA,POM,PVC。当增韧所用的橡胶相形成相互连通网络或使用核壳结构橡胶颗粒时,这类共混物具有超韧性。这种互通的橡胶网络提供了除基体树脂银纹化和屈服之外的“橡胶带”能量耗散机理。
表3是根据ve递增或C∞递减排列的聚合物及其共混物脆-韧转变特性表。脆性基体树脂的ve≅0.1 mmol/cm3,准塑性基体树脂的τ<τc,中间型基体树脂满足上述条件时,它们的共混物都具有超韧特性[12]。

表3 聚合物及其共混物脆-韧转变特性[12]Tab.3Summary of brittle-ductile behavior of polymers and blends
3 结语
橡胶增韧塑料机理的演进是一个发现和继承的不断再认识的过程。虽然Bucknall较早地建立了与橡胶颗粒大小相关的模型,用于解释PA/橡胶体系的增韧机理;但是,他承认设立的模型未考虑橡胶含量变化、温度、应变速率等因素,更重要的是未考虑基体树脂这一重要因素。换言之,有局限性。基于剪切屈服-银纹化理论(剪切带-银纹化理论),Wu Souheng提出的增韧机理定量描述方式较好地将基体树脂的链参数与橡胶形态定量地关联起来,揭示了不同链结构基体树脂及其与橡胶共混物的断裂行为,明确地指出了基体树脂的链参数既控制聚合物/橡胶共混物的本征韧性,又决定了橡胶颗粒的适宜形态。
[1]Ostromislensky I.Process for manufacturing plastic compositions and products obtained thereby:US,1613673[P].1927-01-11.
[2]Newman S,Strella S.Stress-strain behavior of rubber-reinforced glassy polymers[J].J Appl Polym Sci,1965,9(6):2297.
[3]Washiyama J,Kramer E J,Hui C-Y.Fracture mechanism of polymer interface reinforced with block copolymers:transition from chain pullout to crazing[J].Macromolecules,1993,26 (11):2928.
[4]Spurr O K,Niegisch W D.Stress crazing of some amorphous thermoplastics[J].J Appl Polym Sci,1962,6(23):585.
[5]Kambour R P.Optical properties and structure of crazes in transparent glassy polymers[J].Nature,1962,195(4848):1299.
[6]Elias H-G.An introduction to polymer science[M].Weinheim: VCH Verlagsgesellschaft mbH,1997:328.
[7]Kramer E J.Craze fibril formation and breakdown[J].Polymer Engineering and Science,1984,24(10):761.
[8]吴培熙,张留城.聚合物共混改性原理及工艺[M].北京:化学工业出版社,1984:85.
[9]Bucknall C B,Smith R R.Stress-whitening in high-impact polystyrenes[J].Polymer,1965,6(8):437.
[10]孙载坚.塑料增韧[M].北京:化学工业出版社,1982:185.
[11]KambourRP.Areviewofcrazingandfracturein thermoplastics [J].J Polymer Sci:Macromolecular Reviews,1973,7(1):1.
[12]Wu Souheng.Control of intrinsic brittleness and toughness of polymers and blends by chemical structure:a review[J].Polymer International,1992,29(3):229.
[13]Wellinghoff S T,Baer E.Microstructure and its relationship to deformation processes in amorphous polymer glasses[J].J Appl Polym Sci,1978,22(7):2025.
[14]Wu Souheng.Chain structure,phase morphology,and toughness relationships in polymers and blends[J].Polymer Engineering and Science,1990,30(13):753.
[15]Kramer E J.In polymer compatibility and incompatibility: principles and practice[M].New York:MMI Press,Harwood Acad Publ,1982:1.
[16]Keskkula H,Kim H,Paul D R.Impact modification of styreneacrylonitrilecopolymersbymethylmethacrylategraftedrubbers [J].Polymer Engineering and Science,1990,30(21):1373.
[17]Dharmarajan N,Datta S.Toughening styrene maleic anhydride copolymers with functionalized ethylene propylene rubbers[J]. Polymer,1992,33(18):3848.
[18]王国全.聚合物共混改性原理与应用[M].北京:中国轻工业出版社,2007:118.
[19]Bucknall C B,Paul D R.Notched impact behavior of polymer blends:Part 1.New model for particle size dependence[J]. Polymer,2009,50(23):5539.
Advance in research on mechanisms for rubber-toughening of plastics
Wang Rongwei
(SINOPEC Shanghai Research Institute of Petrochemical Technology,Shanghai 201208,China)
The mechanisms for deformation behaviors of rubber-toughened plastics are summarized in terms of their evolution,limitations and relationship with the failure behaviors of glassy polymer matrices.The difference between craze and crack is explained in detail using many experimental evidences in literature. In particular,Souheng Wu′s quantitative method is systematically explained,which is derived from this yieldingcrazing competition theory and reveals the quantitative relationship between molecular chain parameters of the matrices and morphological parameters of the dispersed rubber phase.In addition,Wu′s view is emphasized that chain parameters of the matrices control the intrinsic brittleness and toughness of both matrix and its blend with rubber,and determine the favorable morphology of rubber phase as well.
plastics;rubber;toughening;plasticity;brittleness;craze;crack,yielding
TQ 321.2
A
1002-1396(2012)-02-0069-06
2011-09-30。
2011-12-29。
王荣伟,1960年生,博士,毕业于美国Temple大学,现从事聚合物合成及加工的研究。联系电话:(021)68462197转8511;E-mail:wangrw.sshy@ sinopec.com。
(编辑:吴雅荣)
猜你喜欢
石材(2022年3期)2022-06-01
红岩春秋(2022年1期)2022-04-12
原道(2022年2期)2022-02-17
现代塑料加工应用(2021年5期)2021-02-28
理化检验-化学分册(2020年5期)2020-06-15
意林(绘英语)(2018年2期)2018-11-29
中国塑料(2016年1期)2016-05-17
小天使·二年级语数英综合(2015年11期)2015-11-11
中国塑料(2015年2期)2015-10-14
橡胶工业(2015年8期)2015-07-29
