
低血清谷氨酸转肽酶进行性家族性肝内胆汁淤积症23 例临床分析
2012-12-23 04:22刘丽艳王晓红胡锡琪王建设
中国循证儿科杂志 2012年3期
陆 怡 刘丽艳 王晓红 胡锡琪 王建设 俞 蕙
进行性家族性肝内胆汁淤积症(PFIC)是一组严重的胆汁淤积性肝病,为常染色体隐性遗传性疾病。主要是基因突变而造成肝细胞和胆管上皮细胞上各功能蛋白的生成、修饰和调控缺陷导致肝细胞性胆汁淤积。PFIC 通常在新生儿期或1 岁内起病,以肝内胆汁淤积为临床特征,在儿童期或青春期可因肝功能衰竭致死[1]。常见PFIC 有3型,分别由于ATP8B1、ABCB11 和ABCB4 基因缺陷所致的PFICⅠ、Ⅱ和Ⅲ型;其中PFIC-Ⅰ、Ⅱ型血清谷氨酰转肽酶(GGT)水平正常而PFIC-Ⅲ型GGT 水平升高。
低血清GGT 的PFIC 为一种罕见疾病,其发病率尚无确切报道。PFIC-Ⅰ型源于ATP8B1 的基因缺陷所致FIC1蛋白异常,而PFIC-Ⅱ型源于ABCB11 基因突变导致的胆盐输出泵(BSEP)功能异常。除了基因上的差异外,症状上PFIC-Ⅰ和Ⅱ型相似,均有黄疸和瘙痒等表现[2]。国内尚无诊断为低血清GGT 的PFIC 患儿的临床总结报道。本文对复旦大学附属儿科医院(我院)诊断为低血清GGT 的PFIC患儿的临床特征进行分析,以提高对该病的认识。
1 方法
1.1 纳入标准 ①婴儿期以黄疸和(或)瘙痒为主要特征的患儿;②符合胆汁淤积症的诊断标准[3]:若血清总胆红素(TB)<85. 5 μmol·L-1,结合胆红素(DB)>17.1 μmol·L-1;若TB >85.5 μmol·L-1,DB/TB >20%;③胆汁淤积症状持续且血清GGT 从首次就诊起一直维持低水平(<70 U·L-1)≥6 个月;④在我院就诊和随访的患儿;⑤经ATP8B1 和ABCB11 基因全部外显子测序检测。
1.2 排除标准 ①婴儿期常见胆汁淤积的疾病,如胆道闭锁、各种感染性肝病、药物性肝损害、代谢性肝病和Alagille综合征等;②血清总胆汁酸(TBA)水平正常;③血清GGT水平升高的PFIC-Ⅲ型患儿。
1.3 基因分型方法和分组 采用PCR 技术扩增ATP8B1、ABCB11 基因全部外显子并直接测序,对发现的突变进行反向测序验证[4,5]。ATP8B1 外显子突变为PFIC-Ⅰ型组,ABCB11 外显子突变为PFIC-Ⅱ型组,ATP8B1 和ABCB11 外显子未检出突变为未分型组。
1.4 资料截取 对符合纳入标准的患儿调取病史截取以下项目用于分析:①一般情况:性别、出生体重、发病年龄、基因检测结果和家族史;②临床表现:起病、症状、并发症、肝脾情况、实验室检查、影像学检查和肝脏病理学检查等结果;③治疗、随访和预后情况。
2 结果
2.1 一般情况 2004 年1 月至2007 年6 月临床诊断为低血清GGT 的PFIC 患儿共24 例,其中1 例ATP8B1 和ABCB11 基因测序检测均无异常者,进一步基因检测证实为citrin 缺陷所致胆汁淤积症,予以除外,23 例进入分析。男15 例,女8 例。入院时中位年龄6(2 ~36)个月,中位发病年龄为40 d(0 d 至7 个月)。
PFIC-Ⅰ型组9 例,PFIC-Ⅱ型组7 例,未分型组7 例。23 例均未追问到本病的家族史,其中1 例母孕期胆汁酸升高但无黄疸和肝功能损害;6 例患儿(PFIC-Ⅰ型1 例,PFIC-Ⅱ型5 例)父母行基因检测,均检测到与患儿相同的基因变异。除1 例胎龄36 周因宫内窘迫早产(出生体重2 560 g)之外,其余均为足月儿,出生体重均>2 500 g。
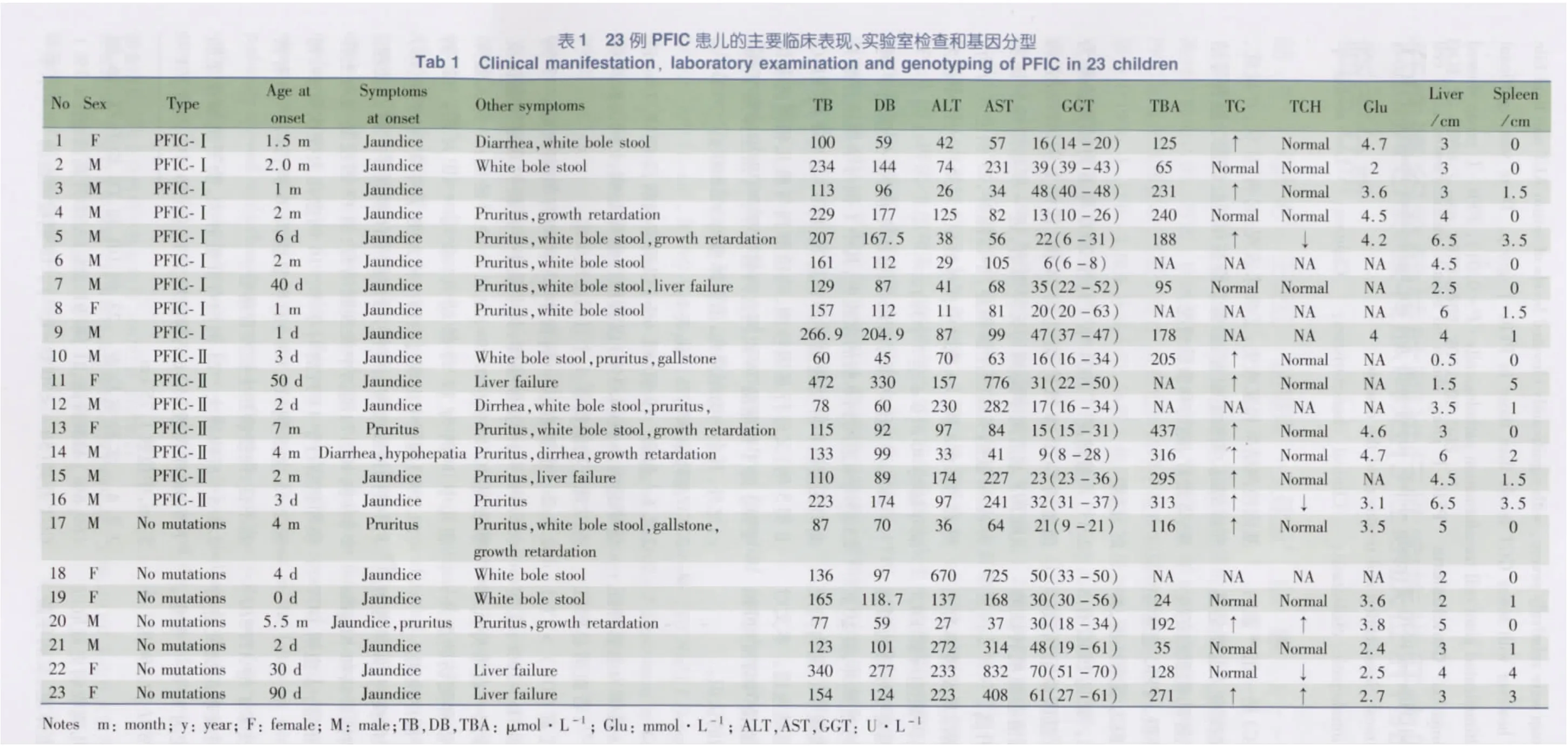
2.2 总体临床特征 见表1。
2.2.1 起病 19 例(82.6%)以黄疸起病,2 例(8.7%)以瘙痒起病,1 例(4.3%)以黄疸伴瘙痒起病,1 例因腹泻检查发现肝功能损害,1 个月后出现黄疸。
2.2.2 主要症状 病程中23 例均有黄疸出现。13 例(56.5%)出现皮肤瘙痒,其中最小5 月龄时观察到瘙抓皮肤表现,10/13 例(76.9%)出现于10 月龄后;10 例无瘙痒患儿中1 例为17 月龄幼儿,余均<10 月龄。12 例(52.2%)病程中出现过白陶土样大便,其中2 例(例2、18)3 月龄内持续白陶土样便,考虑胆道闭锁而手术探查处理。2 例(例10、17)随访中发现胆结石,例17 因反复白陶土样便和胆结石于11 月龄时手术。16 例患儿大便次数多,每天2 ~5 次,3 例(13.0%)有明显腹泻,6 例(26.1%)有营养不良、佝偻病和生长发育落后。
2.2.3 肝脾肿大 23 例均有肝脏肿大,肋下刚触及至肋下6 cm,中位数3.0 cm;脾脏肿大13 例(56.5%),肋下1 ~5 cm,中位数1.75 cm。
?
2.2. 4 实验室检查 23 例DB 和TB 均升高,16 例(69.6%)ALT 升高,21 例(91.3%)AST 升高;2 例(8.7%)GGT 升高(50 ~<70 U·L-1)。18 例检测血清TBA 水平均升高。18 例检测血脂水平,13 例(72. 2%)TG 增高,2 例(11. 1%)TCH 略高,3 例(16.7%)低于正常;6/7 例(85.7%)高密度脂蛋白低于正常。8/15 例(53.3%)空腹血糖<3.9 mmol·L-1,例2 为2.0 mmol·L-1。5 例行血尿串联质谱检测,除1例酰基肉碱轻度增高外均未提示明显异常。
2. 2. 5影像学检查 15/20 例(75.0%)行肝胆同位素排泄显像,提示摄取清除功能差或欠佳,排泄延迟;3 例(15.0%)显示摄取清除功能欠佳,排泄受阻;2 例(10.0%)示摄取清除功能可,排泄受阻。
2.2.6 肝脏病理学检查 16 例有肝脏病理学检查结果,其中12 例为本院肝脏穿刺或外科手术中的肝脏活检标本,3 例外院病理切片本院读片,另有1 例(例18)仅为外院书面病理报告不纳入分析,15 例肝脏病理学检查结果进入分析(表2)。肝活检时平均年龄9.8(2 ~41)个月。光镜下可见肝细胞轻微炎症7 例(46.7%),肝细胞多核巨细胞样转化7 例(46.7%)(图1A),肝细胞胆汁淤积14 例(93.3%),毛细胆管扩张伴胆栓形成14 例(93. 3%)(图1B),汇管区炎症13 例(86.7%),纤维化8 例(53.3%)(图1C),肝硬化2 例(13.3%)(图1D),未发现脂肪变性病例。
2.2.7 治疗、随访和预后 23 例患儿出院时均予以熊去氧胆酸10 ~20 mg·kg-1·d-1治疗,5 例经药物治疗后黄疸消褪至肉眼观察不到。2例分别于5.5 岁和6 岁时因顽固的黄疸和难以控制的瘙痒而行胆汁分流术,分流术后黄疸和瘙痒症状消失。3 例行胆道冲洗术。10 例失访(43.5%),其中2 例出院后失访,8例随访一段时间后失访。13 例随访患儿中7 例(30.4%)死亡(PFIC-Ⅰ、PFIC-Ⅱ和未分型组分别为1、2、4例),其中2 例(例11、22)入院时重症肝炎肝功能衰竭,放弃出院(年龄分别为6 和5 个月),5 例随访中死亡。6 例存活随访患儿平均年龄(78.0 ±13.5)个月,其中4 例身高明显低于同龄儿童(表2)。

表2 23 例PFIC 患儿的肝组织病理学检查、治疗和转归Tab 2 Pathological examination of liver tissue,treatment and outcomes of PFIC in 23 children

图1 PFIC 患儿肝脏病理学检查所见Fig 1 Pathological findings of liver tissue in PFIC children
2.3 PFIC-Ⅰ、PFIC-Ⅱ和未分型组临床特征比较 未分型组ALT、GGT 水平较PFIC-Ⅰ型和PFIC-Ⅱ型组显著升高;PFIC-Ⅱ型组ALT 和TBA 水平显著高于PFIC-Ⅰ型组,余临床特征3 组间差异无统计学意义(表3)。PFIC-Ⅱ型和未分型组肝细胞多核巨细胞转化发生率高于PFIC-Ⅰ型组(4/5 例、3/4 例vs 0/6 例),余病理类型3 组无显著差异。
表3 PFIC-Ⅰ、PFIC-Ⅱ和未分型组临床特征比较[±s,n(%)]Tab 3Comparison of clinical features among PFIC-Ⅰ,PFIC-Ⅱand unclassified groups[±s,n(%)]
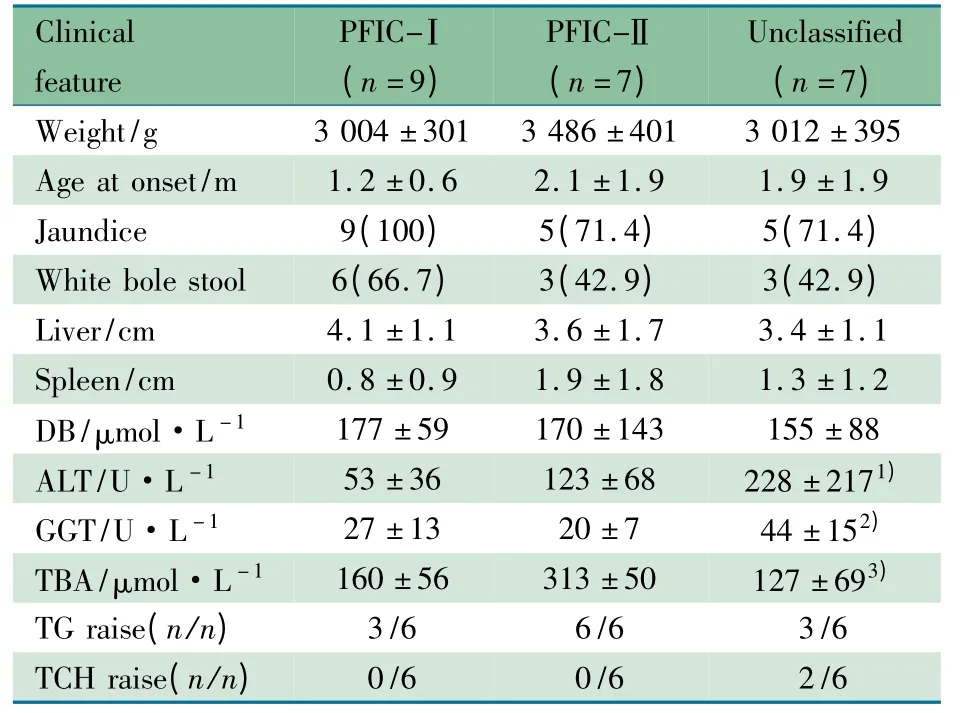
表3 PFIC-Ⅰ、PFIC-Ⅱ和未分型组临床特征比较[±s,n(%)]Tab 3Comparison of clinical features among PFIC-Ⅰ,PFIC-Ⅱand unclassified groups[±s,n(%)]
Notes m:months;vs PFIC-Ⅰand PFIC-Ⅱgroup,1)F=3.799,P=0.040;2)F=5.517,P=0.016;3)F=8.020,P=0.004
Clinical feature PFIC-Ⅰ(n=9)PFIC-Ⅱ(n=7)Unclassified(n=7)Weight/g3 004 ±3013 486 ±401 3 012 ±395 1.9 ±1.9 Jaundice9(100)5(71.4)5(71.4)Age at onset/m 1.2 ±0.6 2.1 ±1.9 3(42.9)Liver/cm4.1 ±1.13.6 ±1.73.4 ±1.1 White bole stool 6(66.7)3(42.9)1.3 ±1.2 DB/μmol·L -1177 ±59170 ±143155 ±88 Spleen/cm 0.8 ±0.9 1.9 ±1.8 228 ±2171)GGT/U·L -127 ±1320 ±744 ±152)ALT/U·L -1 53 ±36 123 ±68 127 ±693)TG raise(n/n)3/66/63/6 TBA/μmol·L -1 160 ±56 313 ±50 2/6 TCH raise(n/n)0/6 0/6
3 讨论
PFIC 是常染色体隐性遗传疾病,其发病机制尚未明确。本组6/23 例患儿父母行ATP8B1 和ABCB11 基因检测,均检测到与患儿相同的突变。PFIC-Ⅰ型是由ATP8B1基因突变影响FIC1 蛋白所致。ATP8B1 位于常染色体18q21-22,编码P 型ATP 酶-FIC1。FIC1 蛋白位于肝细胞毛细胆管膜,在肝内主要由胆管细胞表达[3,6]。目前推断FIC1 蛋白功能异常可间接减少胆管胆汁酸分泌。另外认为ATP8B1 基因突变下调了转录因子法尼酯X 受体(FXR)[7]。FXR 是胆盐感应器,调控胆汁代谢。通过抑制FXR 下调BSEP 表达;还可上调肝细胞胆汁酸合成和回肠肠细胞顶端膜Na+依赖性胆盐转运蛋白(ASBT)表达使胆盐的肠肝循环增加,最终肝内胆汁酸超负荷[8]。PFIC-Ⅰ型胆汁分泌受损还与胆管细胞囊性纤维化跨膜转导调节子(CFTR)表达下调有关,与囊性纤维化样肝外表现有关[3]。PFIC-Ⅱ型源于胆盐排泄泵(BSEP)基因ABCB11 的突变,该基因位于常染色体2q24,编码BSEP 蛋白,BSEP 是肝细胞毛细胆管膜胆盐转运蛋白,属ABC 转运蛋白超家族成员,BSEP 蛋白缺陷致胆盐分泌降低,胆流减少,从而使肝细胞内胆盐积聚,造成严重损伤[8]。
PFIC 临床以黄疸起病为主且呈进行性加重,胆汁淤积所致的高直接胆红素血症可具有反复发作性。特征性的瘙痒症状使患儿非常痛苦,且通过随访发现部分患儿虽然黄疸消褪但瘙痒依旧。喂养困难、脂肪吸收障碍所致的难治性脂肪泻使体重增长和发育缓慢。本组病例临床资料显示,PFIC 起病较早(中位发病年龄40 d),但从出生体重看,患儿宫内发育不受影响,而生后可有生长发育落后,这可能与其发病机制有关。主要临床症状因年龄增长和治疗而有改变,婴儿期(尤其6 月龄内)以黄疸和白陶土样便为主;10 月龄后瘙痒症状愈发明显,1 岁后生长发育落后逐步显现。以上临床特征尤其是黄疸和瘙痒在本组患儿中表现突出,生长发育落后随访中较明显。
文献报道PFIC 的继发表现较多[9],包括脂溶性维生素缺乏症如佝偻病,骨龄延迟,干眼症,凝血功能障碍,维生素E 吸收障碍所致的神经肌肉病变;此外亦可并发肝肿瘤、胆囊结石、肝纤维化门脉高压症和心血管疾病等。其原因可能是由于循环中的高浓度胆盐所致,另外也有报道PFIC 患儿存在脂质代谢异常。本组除2 例随访中发现胆结石外缺少其他明显的继发表现,考虑与随访时间不够长,病例数不够多有关。
本组15/23 例肝活检病理资料可见肝细胞炎症较轻,7例出现肝细胞巨细胞样转化,14 例存在肝细胞胆汁淤积和毛细胆管扩张伴胆栓形成,汇管区炎症(13 例)多于肝细胞炎症(7 例)。文献报道在年长患儿中有胆管损伤导致的胆管缺乏,但本组未有相关病理表现,考虑与活检年龄多≤20月龄有关。文献报道典型的纤维化可始于婴儿期,本组8例在1 岁内出现纤维化并有2 例达肝硬化程度。以上病理结果提示低血清GGT 的PFIC 患儿以肝细胞和胆管内胆汁淤积为主要的肝脏组织学表现,其次为汇管区炎症,但肝细胞炎症不明显;另外肝脏纤维化出现较早且易发展成肝硬化。文献报道[2,10,11]纤维化的发展速度高度变异,而且与疾病症状严重程度无显著联系。本组资料显示,PFIC-Ⅱ型组纤维化与肝硬化比例最高,而未分型组纤维化病例最少,但未分型组的死亡最多,故肝脏纤维化与死亡关系并不平行,与文献报道相符。3 组肝细胞多核巨细胞转化比例差异有统计学意义,其他病理类型如肝细胞炎症、胆汁淤积、胆栓形成、汇管区炎症、纤维化和肝硬化比例无显著差异,考虑可能与样本量较小有关,明确结论仍需扩大样本量进一步证实。
有文献报道[12]低血清GGT 的PFIC 临床诊断患儿行ATP8B1、ABCB11 基因全部外显子测序时,发现15%的患儿无相关突变,本组资料有30%(7/23 例)的临床诊断病例未发现有意义的基因变异,明显高于文献报道。考虑与种族差异或可能其中PFIC 的诊断有待进一步明确,如本组中的2 例(例19、21)TBA 水平不是很高的病例,不排除胆汁酸合成缺陷病的可能。Pawlikowska 等[9]报道血清ALT 或AST 升高、血浆白蛋白降低、外周血WBC 升高在低血清GGT 的PFIC-Ⅰ型和PFIC-Ⅱ型中差异有统计学意义,腹泻症状两组间差异亦有显著统计学意义。本组资料显示,表3 中3 组ALT、GGT 和TBA 水平差异有统计学意义(P <0.05),而起病年龄、起病症状、肝脾肿大程度、黄疸程度、血脂改变3 组间无显著差异(P >0.05);而血浆白蛋白、WBC计数因患儿的多次就诊检查、支持治疗和感染等情况不同未列入统计。本组资料分析发现,PFIC-Ⅱ型组TBA 水平明显高于PFIC-Ⅰ型组,而血清GGT 水平最低。
PFIC-Ⅰ型最初的典型表现是时轻时重的黄疸,但随着病情进展,黄疸可持续不退,无显著升高的转氨酶。PFIC-Ⅱ型的最初表现和进展可能更加严重,出生最初几个月即有持续性黄疸,出生1 年内就可能进展为肝功能衰竭,肝脏的结构因小叶和肝门纤维化以及炎症改变而更加复杂,有更高的转氨酶水平。本组资料有类似发现,但未发现起病年龄方面的差异,从转氨酶升高以及死亡情况看未分型组的病情更重,其次是PFIC-Ⅱ型,其TBA 升高尤其明显;而PFIC-Ⅰ型未发现肝细胞多核巨细胞样改变。本组资料样本量较小,且失访率较高,尚有待进一步探讨。
目前PFIC 的治疗包括:药物治疗,部分胆汁分流术,肝移植等治疗措施。药物治疗是本病最初的治疗策略,包括熊去氧胆酸、苯巴比妥和利福平等,其中熊去氧胆酸疗效相对确切,本组患儿均予熊去氧胆酸10 ~20 mg·kg-1·d-1治疗。长期治疗也许可以延缓肝硬化的进展,推迟肝移植的时间。Jacquemin 等[13]采用熊去氧胆酸20 ~30 mg·kg-1·d-1治疗PFIC 患儿(36 例GGT 水平正常,13 例GGT水平升高)2 ~4 年后ALT 水平显著降低,营养状态改善,肝功能生化检查40%病例正常,30%病例改善,无明显不良反应。本组PFIC-Ⅱ型和未分型组各1 例患儿行胆汁分流术,术后黄疸和瘙痒症状消失,具一定疗效。20 世纪80年代,肝移植被认为是唯一可能治愈该病的治疗措施。但随着研究的进一步深入,发现ATP8B1 基因除了在胆管上皮表达外,还可在小肠、胰腺和肺等上皮细胞表达,肝移植后恢复了胆盐分泌,但不能缓解肝外症状。部分PFIC-Ⅰ型患儿的表现,如腹泻、肝脂肪变性和身高矮小在成功实施胆汁分流术后或肝移植后非但不改善反而恶化[14]。由于严重的肝脂肪变性和(或)脂肪性肝炎,进而逐渐发展至肝硬化,甚至需要再次肝移植[15]。肝移植不能治愈PFIC-Ⅰ型患儿,而PFIC-Ⅱ型可受益于肝移植,但增加了肝癌风险,15%发生肝癌或胆管癌[16]。本组患儿均未行肝移植手术,无法评估其疗效。进一步基因分型有助于治疗方案的选择,也许将来PFIC 患儿可应用细胞、基因或特异性靶位治疗(如FXR 诱导剂和分子伴侣药物)。
PFIC 预后不良,病死率较高,随访过程中可因病情恶化而致肝功能衰竭死亡。本组病例随访到的病死率为30.4%(7/23 例),10 例(43.5%)因家长放弃治疗、路途遥远随访不便和经济因素等失访,其中的死亡病例尚无法统计,本组病例的实际病死率可能更高。
低血清GGT 的PFIC 是渐进性的,其预后除受突变基因的类型和严重程度影响外,还与是否得到恰当干预有关,所有类型的PFIC 如果不经治疗在儿童期将是致命的。本组资料也显示了其难治性和高病死率。本组资料总结以期帮助临床医师识别和区分低血清GGT 的PFIC,以指导临床治疗。
[1]Jacquemin E. Progressive familial intrahepatic cholestasis.Genetic basis and treatment. Clin Liver Dis,2000,4(4):753-763
[2]Vanmil SW,Houwen RH,Klomp LW. Genetics of intrafamilial cholestasis syndromes.J Med Genet,2005,42:449-463
[3]Demeilliers C, Jacquemin E, Barbu V, et al. Altered hepatobiliary gene expressions in PFIC1:ATP8B1 gene defect is associated with CFTR downregulation. Hepatology,2006,43(5):1125-1134
[4]Liu LY,Wang XH,Wang ZL,et al. Characterization of ATP8B1 gene mutations and a hot-linked mutation found in Chinese children with progressive intrahepatic cholestasis and low GGT.J Pediatr Gastroenterol Nutr,2010,50(2):179-183
[5]Liu LY,Wang ZL,Wang XH,et al.ABCB11 gene mutations in Chinese children with progressive intrahepatic cholestasis and low gamma glutamyltransferase. Liver Int,2010,30(6):809-815
[6]Paulusma CC,Groen A,Kunne C,et al. Atp8b1 deficiency in mice reduces resistance of the canalicular membrane to hydrophobic bile salts and impairs bile salt transport.Hepatology,2006,44(1):195-204
[7]Cai SY,Gautam S,Nguyen T,et al. ATP8B1 deficiency disrupts the bile canalicular membrane bilayer structure in hepatocytes,but FXR expression and activity are maintained.Gastroenterology,2009,136(3):1060-1069
[8]Hori T,Nguyen JH,Uemoto S.Progressive familial intrahepatic cholestasis.Hepatobiliary Pancreat Dis Int,2010,9(6):570-578
[9]Pawlikowska L,Strautnieks S,Jankowska I,et al. Differences in presentation and progression between severe FIC1 and BSEP deficiencies.J Hepatol,2010,53(1):170-178
[10]Harris MJ,Le Couteur DG,Arias IM. Progressive familial intrahepatic cholestasis:genetic disorders of biliary transporters.J Gastroenterol Hepatol,2005,20(6):807-817
[11]Kurbegov AC,Setchell KD,Haas JE,et al.Biliary diversion for progressive familial intrahepatic cholestasis:improved liver morphology and bile acid profile. Gastroenterology,2003,125(4):1227-1234
[12]Davit-Spraul A,Fabre M,Branchereau S,et al. ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC):phenotypic differences between PFIC1 and PFIC2 and natural history.Hepatology,2010,51(5):1645-1655
[13]Jacquemin E,Hermans D,Myara A,et al. Ursodeoxycholic acid therapy in pediatric patients with progressive familial intrahepatic cholestasis.Hepatology,1997,25(3):519-523
[14]Lykavieris P,van Mil S,Cresteil D,et al. Progressive familial intrahepatic cholestasis type 1 and extrahepatic features:no catch-up of stature growth,exacerbation of diarrhea,and appearance of liver steatosis after liver transplantation. J Hepatol,2003,39(3):447-452
[15]Hori T,Egawa H,Takada Y,et al. Progressive familial intrahepatic cholestasis:a single-center experience of livingdonor liver transplantation during two decades in Japan. Clin Transplant,2011,25(5):776-785
[16]Knisely AS,Strautnieks SS,Meier Y,et al. Hepatocellular carcinoma in ten children under five years of age with bile salt export pump deficiency.Hepatology,2006,44(2):478-486
猜你喜欢
基层中医药(2022年6期)2022-10-24
现代仪器与医疗(2022年3期)2022-08-12
传染病信息(2022年2期)2022-07-15
肝博士(2022年3期)2022-06-30
中国典型病例大全(2022年12期)2022-05-13
临床外科杂志(2022年11期)2022-01-01
医学前沿(2021年2期)2021-09-10
昆明医科大学学报(2021年5期)2021-07-22
婚育与健康(2020年10期)2020-12-03
中西医结合肝病杂志(2020年2期)2020-10-27
