
新生儿Prader-Willi 综合征13 例临床表型分析
2012-12-23 04:22詹实娜何玺玉王春枝吴虹林
中国循证儿科杂志 2012年3期
詹实娜 何玺玉 王春枝 杨 尧 王 艳 吴虹林 李 昊
Prader-Willi 综合征(PWS)是一种累及全身多系统的印迹遗传性疾病。国外流行病学调查显示PWS 在新生儿中发病率约为1∶30 000,在全部人口中发病率约为1 ∶50 000[1,2],而病死率达3%。PWS 临床表型因年龄不同而存在差异,新生儿期以重度中枢性肌张力低下和吸吮困难为主要表现,随着年龄增长,可逐渐出现食欲亢进、肥胖、生长迟缓、学习困难、智力低下及行为异常等。虽然目前已有PWS 临床筛查[3]和诊断标准[4],但由于特异性不高,尤其在婴幼儿期发现困难,容易与婴儿型脊髓性肌萎缩、脑瘫等引起肌张力低下的疾病相混淆,确诊仍依赖于分子生物学手段。中国PWS 的文献报道较少,且缺乏临床表型的相关研究。本研究回顾性分析在北京军区总医院附属八一儿童医院(我院)确诊为PWS 新生儿的临床表型特征,以期为PWS 的早期筛查提供帮助。
1 方法
1.1 PWS 诊断流程 参照分别由Gunay-Aygun 等[3]和Holm 等[4]提出的PWS 临床筛查及诊断标准,对婴幼儿期原因不明的反应差(如肌张力低下,少哭、少动,喂养困难等),伴或不伴有性腺发育不良,特征性面容等可疑PWS 病例,以甲基化特异性PCR(MS-PCR)方法确诊。确诊病例进一步以甲基化特异性多重连接依赖性探针扩增技术(MS-MLPA)Me028 试剂盒区分父源性15q11.2-q13 区域缺失和母源性同源二倍体。MS-PCR 诊断操作步骤严格按照EpiTect Bisulfite Handbook 04/2006(QIAGEN)说明书进行;MS-MLPA 严格按照MS-MLPA®DNA METHYLATION QUANTIFICATION PROTOCOL (version 15;20-02-2002,MRC-Holland)说明书进行。MS-PCR 和MS-MLPA 结果见图1 和2。

图1 MS-PCR 诊断图示说明Fig 1 Diagnostic charting description of MS-PCR
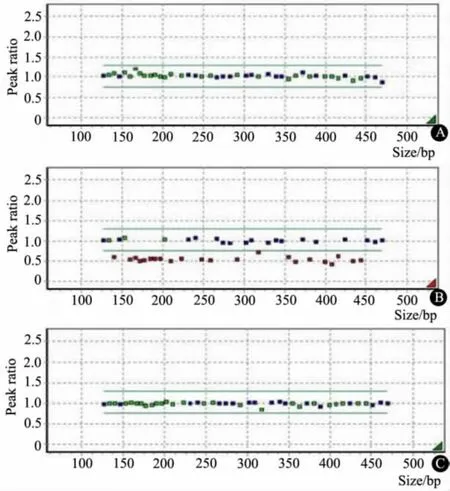
图2 MS-MLPA 诊断图示说明Fig 2 Diagnostic charting description of MS-MLPA
1.2 病例纳入标准 ①我院新生儿病房确诊的PWS 患儿;②汉族;③年龄<28 d。
1.3 资料截取 调取符合纳入标准病例的病史,从中截取以下项目进行分析:(1)一般情况:性别、确诊年龄、出生体重、身长、母亲年龄和胎产式等;(2)基因分型结果;(3)临床表型:提取PWS 诊断标准[4]中新生儿期的临床表型(主要和次要指标各5 项),主要指标包括:①中枢性肌张力,吸吮力;②喂养困难程度,是否需特殊喂养工具;③特征性面容:婴儿期长颅、窄脸、杏仁眼、小嘴、薄上唇、嘴角向下;④生殖器发育情况:男性:阴囊发育,隐睾,与同龄人P5相比阴茎和(或)睾丸大小;女性:生殖器官发育情况;⑤通过高分辨染色体分析(>650 带)或其他细胞或分子诊断方法确诊的15q11-q13 缺失,包括母源性同源二倍体。次要指标:①胎动或婴儿期精神或哭声;②睡眠规律或睡眠中是否存在呼吸暂停;③皮肤颜色;④与同身高人相比手(<P25)和(或)脚(<P10)大小;⑤唾液黏稠度,嘴角是否可见结痂。
2 结果
2.1 一般情况 2009 年8 月至2011 年8 月根据筛选标准疑诊PWS 新生儿34 例,MS-PCR 确诊13 例(38.2%)。9例为父源性15q11.2-q13 区域缺失致病(69.2%),4 例为母源性同源二倍体(30.8%)。男9 例,女4 例,确诊年龄4 ~28d,平均(13 ±10)d;足月儿10 例,早产儿2 例,过期产儿1 例。足月儿出生体重(2 917 ±190)g,头围(34. 7 ±1.7)cm,身长(49.4 ±1.5)cm。孕母≥35 岁9 例。剖宫产11 例,采用剖宫产原因包括:羊水Ⅲ度污染8 例,羊水过多3 例,胎膜早破5 例,异常胎位4 例,宫内窘迫9 例(表1)。

表1 13 例PWS 新生儿的一般临床资料Tab 1 Basic clinical data of 13 neonates with PWS
2.2 临床表型 如表2 所示,4 例母源性同源二倍体患儿均存在中枢性肌张力低下和皮肤色素减退,吸吮缓慢2 例(50. 0%)、哭声微弱3 例(75. 0%)、男性隐睾1 例(25.0%)、女性小阴唇3 例(75.0%)(图3A);均未见特殊面容(图3B)和唾液黏稠,均不需特殊喂养。
9 例父源性15q11.2-q13 区域缺失患儿均存在中枢性肌张力低下、皮肤色素减退和哭声微弱,吸吮缓慢2 例(22. 2%)、男性隐睾7 例(77. 8%)、阴茎短小4 例(44.4%)(图3C)、女性小阴唇1 例(11.1%)、唾液黏稠5例(55.6%)、需特殊喂养7 例(77. 8%)、特殊面容5 例(55.6%)(图3D)。

表2 13 例PWS 新生儿的临床表型Tab 2 The data of clinical phenotypes of 13 neonates with PWS
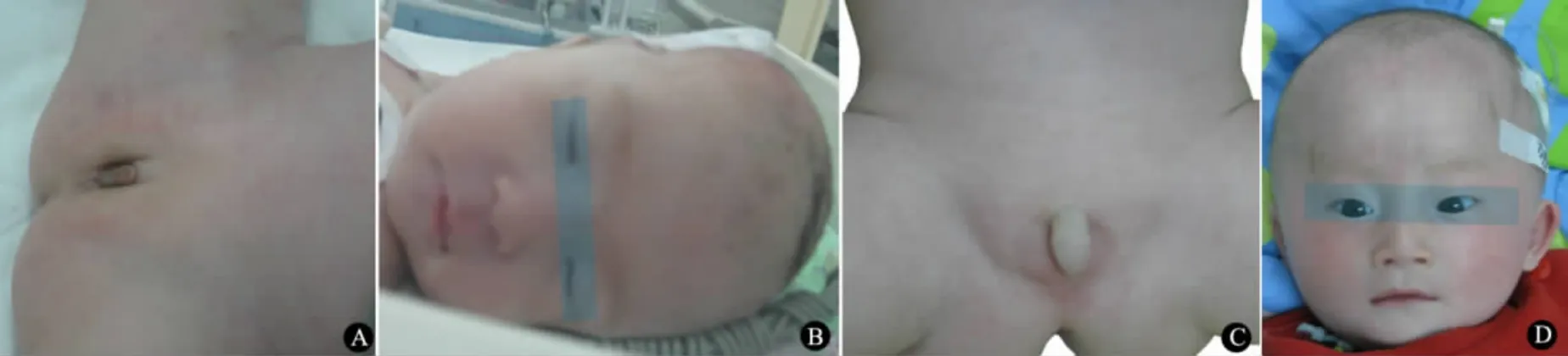
图3 PWS 患儿特殊面容和生殖器发育不全Fig 3 Characteristic facies and abnormal genitals of PWS patients
3 讨论
PWS 是印迹遗传的典型代表,发病机制包括:父源性15q11.2-q13 区域缺失(70% ~75%),母源性同源二倍体(25% ~29%),印迹中心的缺失或突变(<1%)。不同的发病机制所致临床表型和遗传风险存在一定差异。目前研究已鉴别出SNURF-SNRPN,MKRN3,MAGEL2,NDN 及C15orf2 基因为父源性编码多肽的表达基因,其中SNURFSNRPN 是一种混合基因,可编码5 种类型的snoRNAs。最近研究[5]显示SNRPN 下游区的C/D box snoRNAs HBII-85与PWS 表型有密切关系,位于PWS 印迹中心区域,是导致PWS 主要临床表现的基因,但具体控制表型尚不清楚。Rbbp1/Arid4a 和Rbbp1l1/Arid4b 两种Rb-结合蛋白相关基因与印迹中心的印迹调节有关[6]。
PWS 的临床诊断标准由Holm 等[4]于1993 年首先总结提出,新生儿期的表现包括5 项主要指标和5 项次要指标,主要指标每项赋1 分,次要指标每项赋0.5 分,≤3 岁患儿总得分≥5 分(其中4 分来自主要指标)即可作出临床诊断。本研究确诊的13 例PWS 新生儿中,若将主要指标第⑤条包括在内,≥5 分10 例,<5 分3 例。提示该临床诊断标准有可能会漏诊。
2001 年Gunay-Aygun 等[3]对90 例PWS 患者临床表型进行回顾性分析,对上述指标的敏感度和特异度进行了研究,主要指标的敏感度在49%(特征性面容)~98%(婴幼儿期肌张力低下和生长发育迟缓);次要指标的敏感度在37%(睡眠障碍和呼吸暂停)~93%(语言和发音障碍);提出<2 岁患儿可考虑进一步行DNA 分析的指征为肌张力低下和吸吮力差。
文献报道[3]皮肤色素减退发生率在母源性同源二倍体的PWS 患儿中仅为19%,在父源性缺失患儿中为56%。本组13 例PWS 患儿均存在全身性皮肤色素减退,该特征无特异性,但和一般的肤色白有一定差异,即患儿不仅全身肤色白且肤质较薄,弹性较差,红润感不足,毛发色淡;这一皮肤特征也是本组病例最初和最容易被医生关注的表现之一,但与文献报道的阳性率有较大差异。分析原因可能为种族差异,国外报道以白色人种居多,本身皮肤色素淡,PWS 患儿的色素减退表现不明显。中国台湾的一项67 例PWS 患儿的回顾性研究[7]显示皮肤色素减退发生率为82.1%,高于国外文献报道,而低于本组病例,不排除本组病例数较少所致的偏倚,高估了皮肤色素减退的发生率;同时由于该特点存在诊断者主观判断因素且缺乏特异性,确诊后未请皮肤科专业人员评价,亦不排除主观判断的偏倚。此外研究表明PWS 色素减退的发生与编码酪氨酸酶的P基因半合子状态关系密切[8],该基因位于15q11.1-q12,即PWS 印迹区域内。另据文献报道[9]及本组病例特点可见色素减退在母源性同源二倍体中的表现程度较父源性15q11.1-q12 缺失型轻,具体机制尚不清楚,有待进一步深入研究。
本组PWS 新生儿均存在中枢性肌张力低下,与国外98%的报道相近,但每例患儿肌张力低下程度不同,且差别较大,亦存在诊断人员主观判断因素。判断时还可通过患儿的吸吮力量、肌肉松弛度、哭声大小和活动多少等可反映肌张力情况的指标综合判断。研究发现肌张力低下是由于来自伸肌受体传入信号分裂和(或)小脑功能区缺失,导致的由神经支配的梭内肌肌纤维肌梭运动系统失调所引起,进一步影响肌肉肌梭的敏感性,造成被动运动阻力明显减小[10]。故新生儿期肌张力低下容易与神经系统疾病相联系,有文献报道[11]约40% 新生儿期肌张力低下病例是PWS。
本组13 例PWS 新生儿,母源性同源二倍体和父源性15q11.2-q13 区域缺失病例均可观察到肌张力低下和皮肤色素减退。而其他临床表型存在一定的差异,父源性缺失患儿特殊面容、男性隐睾、阴茎短小和唾液黏稠的发生率高于母源性同源二倍体患儿;而女性小阴唇的发生率低于母源性同源二倍体患儿。提示上述临床表型有助于区分PWS 的不同类型,临床上应予以关注。
PWS 患儿大多为足月儿,但由于异常胎位、羊水污染或宫内窘迫等因素常需辅助生产或剖宫产,出生时身长、头围通常正常,与本组病例相符。最近有关超声产前检查PWS 的报道[13]显示,90.9%胎儿出现胎动减少,63.6%宫内臀位及生长受限,36.4%伴羊水过多,而严重生长受限与羊水过多有关(占27.3%)。本组13 例PWS 新生儿均非小于胎龄儿,但出生体重均小于相同胎龄平均体重[14],亦可提示存在一定程度的宫内生长受限。作为次要指标的胎动减少,由于很多孕母缺乏判断经验和标准,孕期检查未按计划进行,故本研究未做统计。孕母高龄者(≥35 岁)为69.2%,提示高龄产妇可能为PWS 的危险因素,与文献报道[15]吻合。
综上,新生儿期皮肤色素减退及中枢性肌张力低下是中国PWS 新生儿普遍存在的特征,可作为进一步行PWS分子诊断的初步筛选标准。母源性同源二倍体和父源性15q11.2-q13 区域缺失患儿的临床表型(特殊面容、生殖器发育不全)有所不同。
PWS 发病率并不低,但由于早期临床表现不典型,且临床医师缺乏对该病的认识,容易造成漏诊和误诊。目前MS-PCR 可以诊断99%的PWS 病例,但临床初步筛选是进行分子诊断的基础。PWS 的临床表现随年龄改变,会面临骨骼发育畸形、饮食过量、语言及运动发育延迟、行为异常、智力低下和身材矮小等问题,相应的对症治疗可明显改善患儿的预后。国外已有通过长期对症治疗及特殊学校教育报道[16]。
[1]VogelsA,Van Den Ende J,Keymolen K,et al. Minimum prevalenve,birth incidence and cause of death for Prader-Willi syndrome in Flander. Eur Jhum Genet,2004,12(3):238-240
[2]Thomson AK,Glasson EJ,Bittles AH. A long-term populationbased clinical and morbidity review of Prader-Willi syndrome in Western Australia. J Intellect Disabil Res,2006,50(Pt 1):69-78
[3]Gunay-Aygun M,Schwartz S,Heeger S,et al. The changing purpose of Prader-Willi syndrome clinical diagnostic criteria and proposed revised criteria. Pediatrics,2001,108(5):E92
[4]Holm VA,Cassidy SB,Butler MG,et al. Prader-Willi syndrome:consensus diagnostic criteria. Pediatrics,1993,91(2):398-402
[5]Sahoo T,del Gaudio D,German JR,et al. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat Genet,2008,40(6):719-721
[6]Wu MY,Tsai TF,Beaudet AL. Deficiency of Rbbp1/Arid4a and Rbbp1l1/Arid4b alters epigenetic modifications and suppresses an imprinting defect in the PWS/AS domain. Genes Dev,2006,20(20):2859-2870
[7]Lin HY,Lin SP,Chuang CK,et al. Genotype and phenotype in patients with Prader-Willi syndrome in Taiwan. Acta Pediatr,2007,96(6):902-905
[8]Spritz RA,Bailin T,Nicholls RD,et al. Hypopigmentation in the Prader-Willi syndrome correlates with P gene deletion but not with haplotype of the hemizygous P allele. Am J Med Genet,1997,71(1):57-62
[9]Dykens EM. Are jigsaw puzzle skills 'spared' in persons with Prader-Willi syndrome?J Child Psychol Psychiatry,2002,43(3):343-352
[10]Marshall S,Teasell R,Bayona N,et al. Motor impairment rehabilitation post acquired brain injury. Brain Inj,2007,21(2):133-160
[11]Nájera N,González L,Pérez Durand J,et al. Small nuclear ribonucleoprotein polypeptide N quantitative methylation analysis in infants with central hypotonia. Pediatr Endocrinol Metab,2011,24(7-8):595-598
[12]Crinò A,Schiaffini R,Ciampalini P,et al. Hypogonadism and pubertal development in Prader-Willi syndrome. Eur J Pediatr,2003,162(5):327-333
[13]Geysenbergh B,De Catte L,Vogels A. Can fetal ultrasound result in prenatal diagnosis of Prader-Willi syndrome?Genet Couns,2011,22(2):207-216
[14]邵肖梅,叶鸿瑁,丘小汕. 实用新生儿学. 第4 版. 北京:人民卫生出版社,2011.954
[15]陆国辉,徐湘民. 临床遗传咨询. 第1 版. 北京:北京大学医学出版社,2007.203
[16]Jin DK. Systematic review of the clinical and genetic aspects of Prader-Willi syndrome. Korean J Pediatr,2011,54(2):55-63
猜你喜欢
全科护理(2022年3期)2022-02-18
中国康复(2021年6期)2021-11-30
特产研究(2021年4期)2021-08-11
新农业(2018年23期)2018-10-17
中国动物检疫(2018年10期)2018-10-16
江苏农业科学(2017年16期)2017-10-27
现代农业科技(2017年8期)2017-06-10
中国现代神经疾病杂志(2017年1期)2017-03-29
中国畜禽种业(2017年5期)2017-01-14
中国神经免疫学和神经病学杂志(2014年5期)2014-05-08
