
HPLC法测定双黄连制剂中连翘苷的含量
2012-12-03 03:35:10代雪平宋汉敏河南省食品药品检验所郑州450003
中国药房 2012年16期
代雪平,宋汉敏(河南省食品药品检验所,郑州 450003)
双黄连类制剂处方由金银花、黄芩、连翘3味药材组成,具有疏风解表、清热解毒的功效,用于外感风热所致的感冒,有口服液、片剂、栓剂、颗粒剂、注射剂等剂型,以口服液和注射剂生产量和使用量最大。由于双黄连类制剂所致不良反应较多[1],对该类药品质量控制的研究也日益增多,其中连翘的主要有效成分连翘苷的测定多采用高效液相色谱(HPLC)法[2]。2010年版《中国药典》(一部)[3](双黄连口服液)及卫生部药品标准[4](双黄连注射液)规定,连翘含量测定项下供试品溶液制备方法为柱层析法,方法操作流程为:精密量取本品15 mL,水浴蒸干,残渣加甲醇2 mL,微热使溶解,加中性氧化铝1 g拌匀,加于中性氧化铝柱(100~120目,10 g,内径2 cm)上,用70%乙醇100 mL洗脱,收集洗脱液,浓缩近干,残渣用50%甲醇溶解后转移至5 mL容量瓶中,用50%甲醇稀释至刻度,摇匀,即得。该方法共10个操作步骤,非常烦琐,且耗时较长(测定周期约2个工作日),不能适应日常检验工作需要。笔者经过分析处方后发现,影响连翘苷测定的主要是黄酮类(以黄芩苷为代表)和有机酸类(以绿原酸为代表)等酚酸性成分,而连翘苷为木脂素类化合物,分子结构中无游离酚羟基或羧基,利用这一结构差异,革除柱层析方法,采用直接稀释后的样品溶液进样,采用弱碱性流动相,使黄芩苷等酚酸类成分成盐后在色谱柱上基本不保留,出峰较快,而连翘苷出峰时间不受影响,可以使连翘苷和其他干扰性成分得到较好分离。现将方法报道如下。
1 仪器与试药
Agilent 1100型HPLC仪,包括G1314 A VWD型紫外检测器、G1312 A型自动进样器、Agilent Chemstation化学工作站(美国Agilent公司);Mettler Toledo十万分之一电子天平(瑞士梅特勒公司)。
连翘苷对照品(中国食品药品检定研究院,批号:110821-200610);双黄连口服液(河南福森药业有限公司,批号:110234;河南天地药业有限公司,批号:20110114);双黄连注射液(河南福森药业有限公司,批号:0910001、100974、1012101、1105051-1、1105091-1、1105101-1、1106351-1、1106361-1、1106371-1、1106381-1、1106401-2、1106421-2、1106431-2、1106441-2、1106451-2、1106571-3、1106581-3、1106591-3);乙腈(德国Merck公司,色谱纯);水为超纯水,其他试剂均为分析纯。
2 方法与结果
2.1 色谱条件
色谱柱:Agilent Zorbax SB-C18柱(250 mm×4.6 mm,5 μm);流动相:乙腈-0.012 mol·L-1醋酸钠溶液(v/v=23∶77);检测波长:277 nm;流速:1 mL·min-1;柱温:35℃;进样量:10 μL。色谱见图1。理论板数按连翘苷计算应不低于4000。由图1可见,在改进后的色谱条件下,供试品溶液中其他成分对连翘苷测定无干扰。
2.2 溶液的制备
2.2.1 对照品溶液的制备 取连翘苷对照品适量,精密称定,加50%甲醇制成每1 mL含0.110 mg的对照品贮备液,精密吸取贮备液10mL,置于50mL容量瓶中,加水稀释至刻度,摇匀,即得。
2.2.2 供试品溶液的制备 双黄连口服液:精密吸取双黄连口服液5 mL,置于25 mL容量瓶中,加无水乙醇稀释至刻度,摇匀,滤过,即得。双黄连注射液:精密吸取双黄连注射液5 mL,置于25 mL容量瓶中,加水稀释至刻度,摇匀,滤过,即得。
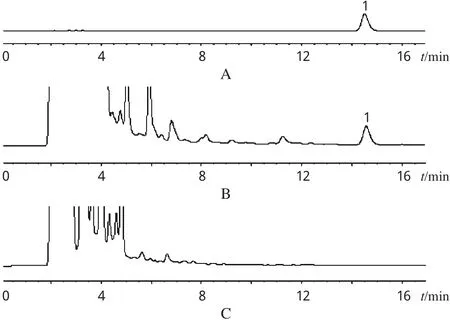
图1 高效液相色谱图Fig 1 HPLC chromatograms
2.2.3 阴性对照溶液的制备 除连翘外,其余各味按处方比例取样,分别照双黄连口服液和注射液“制法”项下规定,制备缺连翘的阴性样品,并按“2.2.2”项下方法制成阴性对照溶液。
2.3 线性关系考察
精密吸取“2.2.1”项下连翘苷对照品贮备液各1、2、5、10、20、30、40、50 μL,分别注入HPLC仪,照“2.1”项下色谱条件测定连翘苷峰面积。以进样量(X,μg)对峰面积(Y)进行线性回归,得回归方程Y=6.6954×102 X+3.9099(r=0.9999)。结果表明,连翘苷进样量在0.11~5.5 μg范围内与峰面积积分值呈良好的线性关系。标准曲线基本过原点,故采用“外标一点法”测定含量。
2.4 精密度试验
精密吸取对照品溶液(浓度为22 μg·mL-1)10 μL,照“2.1”项下色谱条件进样,测定连翘苷色谱峰峰面积,重复测定6次,测定并计算峰面积。结果,连翘苷峰面积RSD=0.5%(n=6),表明仪器精密度良好。
2.5 稳定性试验
取双黄连注射液(批号:0910001)适量,按“2.2.2”项下方法制备供试品溶液,照“2.1”项下色谱条件分别于第0、1、2.5、4、5、6、7、8.5 h进样1次,每次进样10 μL,测定并计算峰面积。结果,连翘苷峰面积RSD=1.7%(n=8),表明供试品溶液在8.5 h内基本稳定。
2.6 重复性试验
取双黄连注射液(批号:0910001)适量,按“2.2.2”项下方法制备供试品溶液,在“2.1”项色谱条件下进样10 μL,测定连翘苷含量,测定6次。结果,连翘苷平均含量为0.116 mg·mL-1,RSD=1.7%(n=6),表明方法重复性较好。
2.7 加样回收率试验
精密量取已知含量的双黄连注射液(批号:0910001,连翘苷含量:0.116 mg·mL-1)适量,共6份,每份5 mL,置于50 mL容量瓶中,分别精密加入连翘苷对照品贮备液(连翘苷含量:0.110 mg·mL-1)5 mL,加水稀释至刻度,摇匀,作为供试品溶液,测定连翘苷含量,计算加样回收率,结果见表1。连翘苷平均加样回收率为98.8%,RSD=0.3%(n=6)。
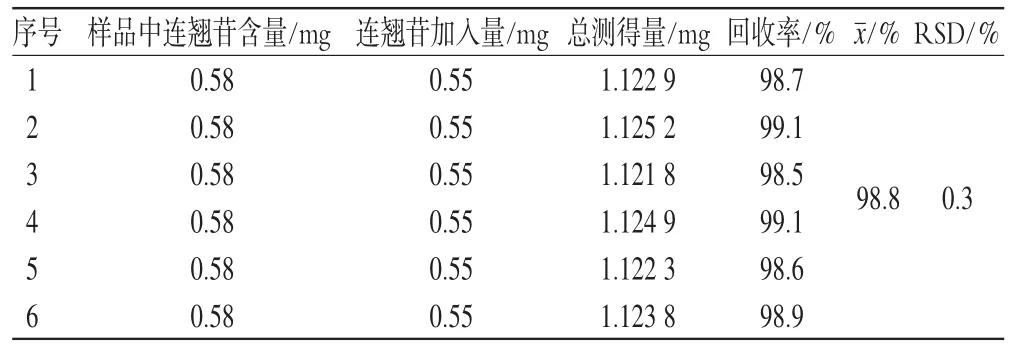
表1 加样回收率试验结果(n=6)Tab 1 Results of recovery tests(n=6)
2.8 样品含量测定
取双黄连口服液和双黄连注射液,按“2.1”项下色谱条件测定连翘苷含量,同时按照2010年版《中国药典》(一部)(双黄莲口服液)及卫生部药品标准(双黄莲注射液)方法测定含量,结果见表2(2种方法测定结果基本一致)。
3 讨论
虽然现代色谱柱填料制造及装填技术的进步已经使液相色谱柱的pH适应范围有了更大的拓展,但十八烷基硅烷键合硅胶填料的化学本质决定了填料在碱性流动相环境下的不稳定性。醋酸钠为弱酸强碱盐,其水溶液呈弱碱性,并在一定的范围内对酸和碱有缓冲作用。笔者考察了不同浓度的醋酸钠溶液的pH值,结合不同浓度的醋酸钠溶液色谱分离效果,最后确定0.012 mol·L-1醋酸钠溶液(pH 6.6)为最优,既可以满足使黄芩苷等酚酸类成分成盐解离而快速洗脱的目的,同时对色谱柱填料的影响最小。
本文采用含醋酸钠溶液流动相替代原国家标准规定的酸性流动相,消除了其他成分对连翘苷在色谱柱分离过程中的干扰,因而简化了连翘苷供试品溶液制备方法,极大地提高了检测效率,改进后的方法连翘苷测定结果与原方法测定结果基本一致,且重复性、稳定性良好,精密度、加样回收率均符合要求,说明改进后的方法完全可以替代原方法。
[1]方世平,杨宝玉.双黄连粉针剂不良反应117例分析[J].中国药房,1998,9(4):176.
[2]聂东东,杨立娟,宋维秋.HPLC法测定双黄连制剂中连翘苷的含量[J].中国药房,2000,11(6):275.
[3]国家药典委员会.中华人民共和国药典(一部)[S].2010年版.北京:中国医药科技出版社,2010:611.
[4]国家药典委员会.中华人民共和国卫生部药品标准-中药成方制剂第十一册[S].WS3-B-2104-96.1996:38.
猜你喜欢
中国民间疗法(2021年8期)2021-07-22 05:53:08
上海计量测试(2020年6期)2021-01-15 03:18:14
今日农业(2020年16期)2020-09-25 03:04:42
基层中医药(2020年2期)2020-07-27 02:45:58
作文新天地(小学版)(2020年5期)2020-06-08 10:29:41
中成药(2018年10期)2018-10-26 03:41:34
CHINESE JOURNAL OF AERONAUTICS(2017年1期)2017-11-21 12:54:14
中成药(2017年5期)2017-06-13 13:01:12
中国病理生理杂志(2015年8期)2015-12-21 12:38:14
意林(2014年10期)2014-07-28 18:17:07
