
Fe、N共掺杂TiO2纳米管阵列的制备及可见光光催化活性
2012-11-30 10:33苏钰丰王梦晔王莹莹林昌健
物理化学学报 2012年3期
吴 奇 苏钰丰 孙 岚 王梦晔 王莹莹 林昌健
(厦门大学化学化工学院化学系,福建厦门361005)
Fe、N共掺杂TiO2纳米管阵列的制备及可见光光催化活性
吴 奇 苏钰丰 孙 岚*王梦晔 王莹莹 林昌健
(厦门大学化学化工学院化学系,福建厦门361005)
应用电化学阳极氧化法结合浸渍和退火后处理制备了Fe和N共掺杂的TiO2纳米管阵列光催化剂,并用场发射扫描电镜(FESEM)、X射线衍射(XRD)、X射线光电子能谱(XPS)和俄歇电子能谱(AES)仪对其进行了表征.结果表明,Fe、N共掺杂对TiO2纳米管阵列的形貌和结构没有明显影响,Fe和N均掺入了TiO2晶格.紫外-可见(UV-Vis)漫反射光谱显示Fe和N共掺杂TiO2纳米管阵列的吸收带边较纯TiO2纳米管阵列和单一掺杂TiO2纳米管阵列红移,可见光吸收增强.以可见光催化降解罗丹明B(RhB)考察了材料的光催化活性,Fe和N共掺杂TiO2纳米管阵列对RhB的降解速率较纯TiO2纳米管阵列和单一掺杂TiO2纳米管阵列明显提高,证明了Fe、N共掺杂产生的协同效应提高了TiO2纳米管阵列在可见光照射下的光催化活性.
Fe和N;共掺杂;TiO2纳米管阵列;罗丹明B;可见光
1 引言
电化学阳极氧化法制得的高度有序的二氧化钛(TiO2)纳米管阵列膜除了具有传统TiO2的催化活性高、价廉、耐腐蚀性强以及无污染1等一系列优点外,还具有比表面积大、光生电子传导速率快、易施加偏压,介电性能优良等特点,2近年来已成为国内外竞相研究的热点之一.然而,由于其和传统的TiO2一样带隙较宽(Eg=3.2 eV),只能吸收波长小于387.5 nm的紫外光,而这部分紫外光(λ<400 nm)仅约占太阳光的4%,在太阳光谱中占绝大多数的可见光部分未得到有效利用,从而限制了其实际应用.因此,开发可见光响应型TiO2纳米管阵列光催化剂成为目前光催化领域科学家所面临的一个重要挑战.已有的研究发现,对TiO2纳米管阵列进行适当的掺杂或表面改性如非金属离子掺杂、3-6金属离子掺杂、7,8贵金属修饰、2,9半导体复合10,11和染料敏化12-14等可以增强TiO2纳米管阵列对可见光的吸收,有效地抑制光生电子和空穴的复合,从而提高其可见光光催化活性.在用于对TiO2进行改性的非金属元素中,N由于具有和O相似的尺寸,能形成亚稳的深能级受主(AX)中心以及较小的电离能而被认为是最有效的掺杂剂.15近年来,一些非金属共掺杂TiO2纳米管阵列的研究工作16,17证实了非金属共掺杂产生的二元协同效应可以提高TiO2纳米管阵列的可见光光催化活性,其对有机污染物的可见光催化效率优于单一掺杂.然而,对TiO2纳米管阵列进行阴阳离子共掺杂鲜有报道.众所周知,对TiO2进行金属离子掺杂可在其晶格中引入新电荷、形成缺陷或改变晶格类型,从而改变TiO2的能带结构,最终改变TiO2的光催化活性.Fe3+具有半充满d电子构型,且离子半径与Ti4+相近,是理想的掺杂金属之一.通过在阳极氧化的电解液中加入Fe3+已经成功制得了Fe掺杂TiO2纳米管阵列.8Isimjan等18在含有K3Fe(CN)6的乙二醇溶液中采用一步阳极氧化的方法成功制得了Fe、C、N共掺杂的TiO2纳米管阵列,有效地提高了TiO2纳米管阵列对可见光的响应.有研究表明,采用不同离子共掺杂,19如阴、阳离子共掺杂20-22等方法不仅可以增大TiO2光催化剂的光吸收范围,而且能够有效地提高其光催化活性.
本工作采用电化学阳极氧化法结合浸渍和退火后处理在纯Ti表面制备Fe和N(Fe-N)共掺杂的TiO2纳米管阵列光催化剂,分析掺杂离子在阵列膜层内的分布.以罗丹明B为目标污染物,考察Fe和 N共掺杂TiO2纳米管阵列的可见光光催化活性,并与纯TiO2纳米管阵列和单一掺杂TiO2纳米管阵列的光催化活性进行对比.
2 实验部分
2.1 实验材料和试剂
工业纯Ti板(纯度>99%)由福建冶金工业研究所提供.所用试剂HF(40%,质量分数,下同)、Fe(NO3)3、NH3·H2O(25%-28%)和罗丹明B(RhB)均为分析纯,且均为国药集团化学试剂有限公司生产.
2.2 Fe、N共掺杂TiO2纳米管阵列的制备
将工业级纯Ti板用水磨砂纸逐级打磨至表面无明显划痕,然后依次用丙酮、无水乙醇、去离子水超声清洗.在室温下,Ti板为阳极,铂片为阴极,分别以0.5%(质量分数)HF水溶液和含有0.1 mol·L-1Fe(NO3)3的0.5%HF水溶液为电解液,在10 V电压下阳极氧化30 min,然后用大量去离子水冲洗样品表面,自然干燥.将样品置于马弗炉中450°C恒温热处理2 h,分别制得纯TiO2纳米管阵列(TiO2NTs)和Fe掺杂TiO2纳米管阵列(Fe-TiO2NTs).将上述Fe掺杂TiO2纳米管阵列样品在1 mol·L-1NH3·H2O溶液中浸泡10 h,取出后立即放入马弗炉中在450°C恒温热处理2 h,制得Fe-N共掺杂TiO2纳米管阵列(Fe-N-TiO2NTs).为了进行对比,将纯TiO2纳米管阵列在1 mol·L-1NH3·H2O溶液中浸泡10 h,取出后在马弗炉中450°C恒温热处理2 h,制得N掺杂TiO2纳米管阵列(N-TiO2NTs).
2.3 Fe、N共掺杂TiO2纳米管阵列的表征
采用日本日立公司S4800场发射扫描电子电镜(FESEM)观察样品的表面形貌并测试膜层厚度.采用荷兰Philips公司Panalytical Xʹpert转靶X射线衍射(XRD)仪测试样品的晶体结构.测试参数为:Cu Kα靶,λ为0.15406 nm,工作电流30 mA,工作电压40 kV,狭缝系统为1 DS-1 SS-0.15 mm RS,以石墨单色器滤波.采用英国VG公司Physical Electrons Quantum 2000 Scanning Esca Microprob光电子能谱仪测定样品的X射线光电子能谱(XPS).Al靶Kα射线,结合能以C 1s(284.8 eV)为基准.采用美国Varian公司Cary 5000型紫外-可见-近红外(UV-Vis-NIR)分光光度计表征样品的光吸收特性.采用美国Perkine-Elemer公司生产的PHI 660俄歇电子能谱(AES)仪检测阵列膜层内的组分及含量.
2.4 TiO2纳米管阵列可见光光催化活性测试
光催化实验是在自制的石英双层夹套反应装置中进行,内层装有罗丹明B(RhB)溶液,外层持续通过冷却水,反应体系的温度控制在30°C.采用500 W卤钨灯作为光源.具体过程为:将TiO2纳米管阵列样品(10 mm×10 mm)放入30 mL、5 mg·L-1RhB水溶液中浸泡30 min,并持续通入纯净空气,使样品与RhB溶液之间达到吸脱附平衡.样品表面距离光源10 cm,样品与光源之间加一滤光片,用以滤去波长λ<420 nm的光.光照的同时开始计时,应用日本岛津公司生产的UV-2100型紫外-可见分光光度计在λ=558 nm处测定光催化过程中RhB的吸光度变化,以此计算RhB的残余浓度.根据一级动力学公式ln(C0/C)=kt(其中C0,C分别为起始和光照t时间后溶液的浓度),线性拟合实验数据可求出光催化降解的表观反应速度常数k,用以判断和比较催化剂的光催化活性.
3 结果与讨论
3.1 SEM和XRD分析
图1为TiO2纳米管阵列和分别经过掺Fe、N以及Fe-N共掺杂改性后的TiO2纳米管阵列的SEM图,图1(a)中的插图为TiO2纳米管阵列的侧面SEM图.从图1(a)可以看出,TiO2膜层表面呈规则的纳米管阵列排布,纳米管直径为40-60 nm,膜层厚度约为400 nm.比较图1(a)与(b)、(c)、(d)可以看出,掺杂后的TiO2纳米管阵列的形貌结构没有明显变化,只是纳米管阵列表面的粗糙程度较未掺杂的TiO2纳米管阵列有所增加.
图2为TiO2纳米管阵列和分别经过掺Fe、N以及Fe-N共掺杂改性后的TiO2纳米管阵列的XRD图.图中所有谱线在25.3°和47.9°处都各有一特征峰,分别为锐钛矿TiO2的(101)和(200)衍射峰(JCPDS No.21-1272),这表明经过450°C热处理后所得到的纳米管阵列膜为锐钛矿相.与未掺杂的TiO2纳米管阵列膜进行比较,发现N掺杂对TiO2晶体结构影响不大,而经过 Fe掺杂后,Fe-TiO2NTs和Fe-NTiO2NTs样品中(101)和(200)衍射峰的强度明显减弱,这一结果与已报道的结果8一致,是由于Fe的掺入抑制了TiO2由无定型向锐钛矿相的转变所致.
3.2 XPS和AES分析
图3(a)为Fe-N共掺杂TiO2纳米管阵列的XPS全谱图.从图中可以看出,制得的样品中含有Ti、O、Fe和N元素.图3(b,c)分别为Fe 2p和N 1s的高分辨XPS谱图.图3(b)中位于710.7和724.5 eV的谱峰分别归属于Fe 2p3/2和Fe 2p1/2,表明Fe元素是以Fe3+的形式23取代了TiO2中部分Ti4+进入了TiO2晶格中,形成Ti-O-Fe网络.图3(c)中位于(402±0.2)eV附近的谱峰归属于吸附的零价态的N,24位于(396±0.2)eV处的XPS峰则归属于β-N(N3-),25这主要是由于TiO2纳米管阵列在由无定型转变为锐钛矿型的过程中,部分N以取代晶格中氧的形式进入TiO2晶格中,形成N-Ti-O网络所致.Fe3+和N3-的掺入所形成的Ti-O-Fe网络和N-Ti-O网络增加了TiO2的活性比表面积和表面缺陷,26,27二者的协同作用有利于拓宽TiO2的光响应范围和提高材料的光催化活性.28

图1 纳米管阵列的SEM图Fig.1 SEM images of nanotube arrays NT:nanotube array;(a)pure TiO2NTs,(b)Fe-TiO2NTs,(c)N-TiO2NTs,(d)Fe-N-TiO2NTs

图2 不同样品的XRD图Fig.2 XRD patterns of different samples
为了确定掺入膜层的Fe元素和N元素在膜层内由表及里的分布情况,测试了Fe-N共掺杂TiO2纳米管阵列的AES谱图,如图4所示.膜层中F元素的含量极低,它是在阳极氧化过程中由HF电解液引入的.在样品表面O和Ti的原子数之比大于2:1,随着阵列膜层深度的增加,O和Ti的原子数之比逐渐接近2:1.这主要是由于纳米管阵列表面含有吸附态的O的缘故,膜层越深,吸附态的O越少,O/Ti比值趋于2:1.同时可以看出,膜层中Fe的含量很少而且也由表及里逐渐降低,这表明Fe元素主要存在于纳米管表面和一定深度的膜层内.然而,N元素的含量随着膜层深度的增加,先快速增加而后缓慢增加,并趋于稳定,这表明N元素较均匀地分布在整个TiO2纳米管阵列膜层中.
3.3 UV-Vis吸收光谱
图5比较了纯TiO2和Fe掺杂、N掺杂以及Fe-N共掺杂TiO2纳米管阵列在200-800 nm波长范围内的光吸收.插图为根据公式(αhν)2=A(hν-Eg) (式中,α为吸收系数,A为与材料有关的常数,h为普朗克常数,ν为光的频率,Eg为禁带能量)29计算得到的(αhν)1/2

图3 Fe-N共掺杂TiO2纳米管阵列的XPS谱图Fig.3 XPS patterns of Fe-N codoped TiO2nanotube arrays(a)XPS survey spectrum;(b)Fe 2p;(c)N 1s

图4 Fe-N共掺杂TiO2纳米管阵列膜层各种元素的AES图Fig.4 AES of the different elements in Fe-N codoped TiO2 nanotube array film
-hν的关系曲线.可以看出,纯TiO2纳米管阵列的主要吸收波长小于380 nm的紫外光,对应于锐钛矿型TiO2的本征吸收,而其在可见光范围内小的波动峰是由于TiO2纳米颗粒捕获电荷载流子的吸收叠加造成的.26与TiO2纳米管阵列的光吸收进行比较,Fe、N掺杂以及Fe-N共掺杂TiO2纳米管阵列光吸收带边均发生了不同程度的红移,在可见光区的光吸收增强.从(αhν)1/2-hν关系图中可以得到TiO2纳米管阵列、N掺杂、Fe掺杂和Fe-N共掺杂TiO2纳米管阵列的带隙分别为3.26、3.03、2.93和2.73 eV,即带隙顺序为TiO2NTs>N-TiO2NTs>Fe-TiO2NTs>Fe-N-TiO2NTs,进一步说明Fe和N成功地掺入了TiO2晶格,增强了TiO2纳米管阵列对可见光的响应.Fe掺杂TiO2纳米管阵列吸收带边红移是由于Fe3+的3d轨道上的电子能被可见光激发,电荷在3d轨道与TiO2的导带或价带之间转移造成440 nm附近的吸收;30而550 nm附近的吸收与Fe3+的d-d跃迁(2T2g→2A2g,2T1g)或相互作用的Fe离子之间的电荷转移(Fe3++Fe3+→Fe4++Fe2+)有关.31N掺杂TiO2纳米管阵列中,N元素2p轨道的电子态会与O元素2p轨道的电子态混合,使TiO2带隙变窄,光响应红移到可见光区,32同时N掺杂还会在TiO2表面形成较稳定的氧空位,可增强TiO2的可见光响应.33在三种掺杂TiO2纳米管阵列中,由于Fe-N共掺杂表现出协同作用,因而Fe-N共掺杂TiO2纳米管阵列的可见光响应最为显著.

图5 不同样品的紫外-可见吸收光谱Fig.5 UV-Vis diffuse reflectance spectra of different sampleshv:photo energy;The inset is the estimated band gap by Kubelka-Munk function.
3.4 光催化活性
不同掺杂的光催化剂可见光光催化降解RhB的ln(C0/C)-t关系图示于图6.相应的反应速率常数列于表1.由图可以看出,RhB在可见光照射下存在一定的自降解,单一的Fe或N掺杂TiO2纳米管阵列的光催化活性均高于纯TiO2纳米管阵列;Fe-N共掺杂纳米管阵列的光催化活性最高,其反应速率常数较纯TiO2纳米管阵列提高了约1倍.四种催化剂光催化活性的顺序是:TiO2<Fe-TiO2<N-TiO2<Fe-N-TiO2.
由于可见光下RhB可发生光敏化,34因而有一定的自降解.当以纯TiO2纳米管阵列为催化剂时,由于TiO2不能被可见光激发,因此TiO2纳米管阵列对RhB的降解是因RhB的光敏化所致.RhB吸附在TiO2纳米管阵列表面的Ti4+位上,35在可见光下RhB被激发,受激电子注入到TiO2导带并被吸附在表面的溶解氧捕获生成超氧自由基,进而将RhB氧化降解.反应过程如下:

图6 不同样品光催化降解RhB的动力学曲线Fig.6 RhB photodegradation kinetic curves of the different samples
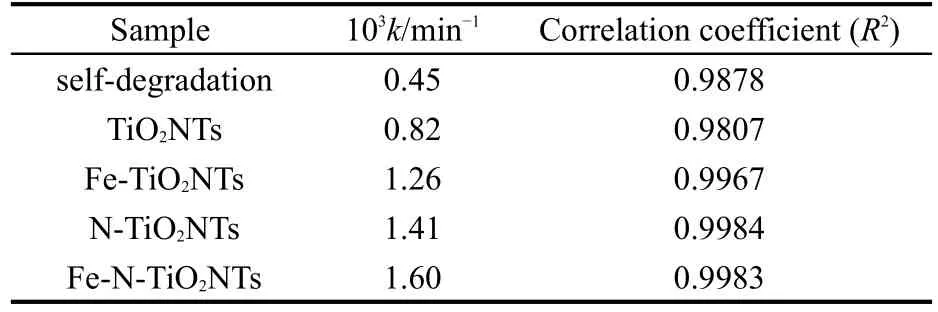
表1 不同样品在可见光照射下降解罗丹明B的一级表观速率常数(k)Table 1 First-order apparent rate constants(k)of different samples for RhB photodegradation under visible light irradiation
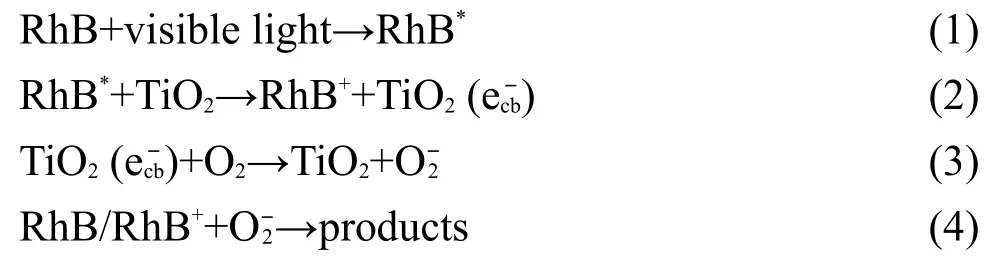
上式中RhB*是光照条件下罗丹明B生成的活性中间体,e-
cb表示位于TiO2导带上的电子.当以Fe掺杂TiO2纳米管阵列为催化剂时,由于Fe3+/Fe4+能级高于锐钛矿相TiO2的价带底,36,37Fe掺杂TiO2能够吸收波长大于400 nm的光.在可见光下,3d电子从Fe3+中心被激发到TiO2导带,在Fe3+/Fe4+能级上留下Fe4+.导带上的受激电子还原吸附在表面的溶解氧产生O-2可降解RhB,而Fe4+能够将溶液中的OH-氧化成羟基自由基·OH,从而进一步降解RhB.同时,由于Fe3+可以和带有羧酸基的有机物分子形成配位化合物,38因而RhB也可以吸附在Fe-TiO2表面的Fe3+位上被Fe4+氧化成Fe3+.39反应过程如下:

4 结论
采用阳极氧化法先制得Fe掺杂TiO2纳米管阵列,再通过在NH3·H2O溶液中浸渍和随后的热处理制备出Fe-N共掺杂TiO2纳米管阵列.Fe和N的掺入使TiO2纳米管阵列的起始带边发生了明显的红移,对可见光的吸收随之增强.可见光降解RhB实验表明,掺杂提高了TiO2纳米管阵列的光催化效率, Fe-N共掺杂产生的协同效应共同提高了TiO2纳米管阵列在可见光下降解RhB的效率,表现出最好的光催化活性.
(1) Zhang,X.R.;Lin,Y.H.;Zhang,J.F.;He,D.Q.;Wang,D.J. Acta Phys.-Chim.Sin.2010,26,2733. [张晓茹,林艳红,张健夫,何冬青,王德军.物理化学学报,2010,26,2733.]
(2) Xie,K.P.;Sun,L.;Wang,C.L.;Lai,Y.K.;Wang,M.Y.;Chen, H.B.;Lin,C.J.Electrochim.Acta 2010,55,7211.
(3) Lu,N.;Quan,X.;Li,J.Y.;Chen,S.;Yu,H.T.;Chen,G.H. J.Phys.Chem.C 2007,111,11836.
(4) Park,J.H.;Kim,S.;Bard,A.J.Nano Lett.2005,6,24.
(5) Tang,X.H.;Li,D.Y.J.Phys.Chem.C 2008,112,5405.
(6) Vitiello,R.P.;Macak,J.M.;Ghicov,A.;Tsuchiya,H.;Dick,L. F.P.;Schmuki,P.Electrochem.Commun.2006,8,544.
(7) Liu,H.J.;Liu,G.G.;Zhou,Q.X.J.Solid.State.Chem.2009, 182,3238.
(8) Sun,L.;Li,J.;Wang,C.L.;Li,S.F.;Chen,H.B.;Lin,C.J.Sol. Energy Mater.Sol.Cells 2009,93,1875.
(9) Mohapatra,S.K.;Kondamudi,N.;Banerjee,S.;Misra,M. Langmuir 2008,24,11276.
(10) Wang,C.L.;Sun,L.;Yun,H.;Li,J.;Lai,Y.K.;Lin,C.J. Nanotechnology 2009,20,295601.
(11)Hou,Y.;Li,X.Y.;Zhao,Q.D.;Quan,X.;Chen,G.H.Adv. Funct.Mater.2010,20,2165.
(12) Zhu,K.;Neale,N.R.;Miedaner,A.;Frank,A.J.Nano Lett. 2006,7,69.
(13) Wang,J.;Lin,Z.Q.Chem.Mater.2009,22,579.
(14)Ye,M.D.;Xin,X.K.;Lin,C.J.;Lin,Z.Q.Nano Lett.2011,11, 3214.
(15) Park,C.H.;Zhang,S.B.;Wei,S.H.Phys.Rev.B 2002,66, 073202.
(16) Chen,X.Q.;Su,Y.L.;Zhang,X.W.;Lei,L.C.Chin.Sci.Bull. 2008,53,1983.[陈秀琴,苏雅玲,张兴旺,雷乐成.科学通报,2008,53,1983.]
(17) Zhou,X.S.;Peng,F.;Wang,H.J.;Yu,H.;Yang,J.Electrochem. Commun.2011,13,121.
(18) Isimjan,T.T.;Ruby,A.E.;Rohani,S.;Ray,A.K. Nanotechnology 2010,21,055706.
(19) Shen,Y.F.;Xiong,T.Y.;Du,H.;Jin,H.Z.;Shang,J.K.;Yang, K.J.Sol-Gel.Sci.Technol.2009,50,98.
(20) Li,X.;Chen,Z.M.;Shi,Y.C.;Liu,Y.Y.Powder Technol.2011, 207,165.
(21)Yang,X.X.;Cao,C.D.;Erickson,L.;Hohn,K.;Maghirang,R.; Klabunde,K.Appl.Catal.B:Environ.2009,91,657.
(22) Sun,X.J.;Liu,H.;Dong,J.H.;Wei,J.Z.;Zhang,Y.Catal. Lett.2010,135,219.
(23) Tong,T.Z.;Zhang,J.L.;Tian,B.Z.;Chen,F.;He,D.N. J.Hazard.Mater.2008,155,572.
(24) Saha,N.C.;Tompkins,H.G.J.Appl.Phys.1992,72,3072.
(25)Ao,Y.H.;Xu,J.J.;Fu,D.G.;Yuan,C.W.J.Hazard.Mater. 2009,167,413.
(26) Lu,W.L.;Shi,L.Y.;Xie,X.F.;Zhang,J.P.;Li,X.L.Journal of Shanghai University(Natural Science)2004,10,289. [陆文璐,施利毅,谢晓峰,张剑平,李希玲.上海大学学报(自然科学版),2004,10,289.]
(27)Yu,X.B.;Wang,G.H.;Luo,Y.Q.;Chen,X.H.;Zhu,J.Journal of Shanghai Teachers University(Natural Science)2000,29, 75.[余锡宾,王桂华,罗衍庆,陈秀红,朱 建.上海师范大学学报(自然科学版),2000,29,75.]
(28) Tao,J.J.;Zhang,Y.F.;Zhang,L.F.;Yao,H.;Liu,Y.H.Journal of Functional Materials 2010,42,824. [陶金建,章宇飞,张廉奉,姚 皓,刘艳华.功能材料,2010,42,824.]
(29)Wu,D.Y.;Long,M.C.;Cai,W.M.;Chen,C.;Wu,Y.H. J.Alloy.Compd.2010,502,289.
(30) Umebayashi,T.;Yamaki,T.;Itoh,H.;Asai,K.J.Phys.Chem. Sol.2002,63,1909.
(31) Zhu,J.F.;Chen,F.;Zhang,J.L.;Chen,H.J.;Anpo,M. J.Photochem.Photobiol.A-Chem.2006,180,196.
(32)Asahi,R.;Morikawa,T.;Ohwaki,T.;Aoki,K.;Taga,Y.Science 2001,293,269.
(33) Ihara,T.;Miyoshi,M.;Iriyama,Y.;Matsumoto,O.;Sugihara,S. Appl.Catal.B 2003,42,403.
(34) Ma,Y.;Yao,J.N.J.Photochem.Photobiol.A-Chem.1998,116, 167.
(35) Li,Y.X.;Lu,G.X.;Li,S.B.J.Photochem.Photobiol.A-Chem. 2002,152,219.
(36) Ma,Y.;Zhang,X.T.;Guan,Z.S.;Cao,Y.A.;Yao,J.N. J.Mater.Res.2001,16,2928.
(37) Zhu,J.F.;Zheng,W.;He,B.;Zhang,J.L.;Anpo,M.J.Mol. Catal.A-Chem.2004,216,35.
(38) Litter,M.I.;Baumgartner,E.C.;Urrutia,G.A.;Blesa,M.A.; Environ.Sci.Technol.1991,25,1907.
(39) Cao,J.L.;Wu,Z.C.;Cao,F.H.;Zhang,J.Q.J.Inorg.Mater. 2007,22,514.[曹江林,吴祖成,曹发和,张鉴清.无机材料学报,2007,22,514.]
(40) Ma,Y.F.;Zhang,J.L.;Tian,B.Z.;Chen,F.;Wang,L.Z. J.Hazard.Mater.2010,182,386.
(41)Wu,Y.M.;Xing,M.Y.;Tian,B.Z.;Zhang,J.L.;Chen,F. Chem.Eng.J.2010,162,710.
November 14,2011;Revised:December 15,2011;Published on Web:December 23,2011.
Preparation and Visible Light Photocatalytic Activity of Fe-N Codoped TiO2Nanotube Arrays
WU Qi SU Yu-Feng SUN Lan*WANG Meng-Ye WANG Ying-Ying LIN Chang-Jian
(Department of Chemistry,College of Chemistry and Chemical Engineering,Xiamen University,Xiamen 361005, Fujian Province,P.R.China)
Fe-N codoped TiO2nanotube arrays were fabricated by anodization of Ti,followed by wet immersion and annealing post-treatment.The dopedTiO2nanotube arrayphotocatalystswere characterized by field-emission scanning electron microscopy(FESEM),X-ray diffraction(XRD),X-ray photoelectron spectroscopy(XPS),and Auger electron spectroscopy(AES).The results indicated that Fe and N dopants had almost no effect on the morphology and structure of TiO2nanotube arrays,and that Fe and N were doped into the TiO2lattice.UV-Vis diffuse reflectance spectra showed that the absorption band edge of Fe-N codoped TiO2nanotube arrays exhibited a red shift compared with that of pure TiO2nanotube arrays and Fe-or N-doped TiO2nanotube arrays.The photocatalytic activity of Fe-N codoped TiO2nanotube arrays was evaluated by their ability to degrade rhodamine B under visible light irradiation.The degradation rate of rhodamine B over Fe-N codoped TiO2nanotube arrays was obviously higher than that over pure TiO2nanotube arrays and Fe-or N-doped TiO2nanotube arrays,which is attributed to the synergistic effect of the Fe and N codopants.
Fe and N;Codoping;TiO2nanotube array;Rhodamine B;Visible light
10.3866/PKU.WHXB201112231
O644
∗Corresponding author.Email:sunlan@xmu.edu.cn;Tel:+86-10-2184655.
The project was supported by the National Natural Science Foundation of China(51072170,21021002),Natural Science Foundation of Fujian Province,China(2011J01057),and National Foundation for Fostering Talents of Basic Science,China(J1030415).
国家自然科学基金(51072170,21021002),福建省自然科学基金(2011J01057)和国家基础科学人才培养基金(J1030415)资助项目
猜你喜欢
四川地质学报(2022年2期)2022-07-08
矿产勘查(2020年8期)2020-12-25
原子与分子物理学报(2020年5期)2020-03-17
无线互联科技(2019年15期)2019-11-07
分析化学(2018年12期)2018-01-22
无机盐工业(2017年6期)2017-03-11
中国纤检(2017年1期)2017-03-07
中国塑料(2016年8期)2016-06-27
中国塑料(2016年12期)2016-06-15
功能材料(2016年1期)2016-05-17
