
氢键复合物中键长变化与振动频率移动相关性重访
2012-11-30 10:33:40王伟周
物理化学学报 2012年3期
张 愚 马 宁 王伟周
(洛阳师范学院化学化工学院,河南洛阳471022)
氢键复合物中键长变化与振动频率移动相关性重访
张 愚 马 宁 王伟周*
(洛阳师范学院化学化工学院,河南洛阳471022)
X―H···Y(Y为电子供体)型氢键形成时,X―H键长伸长或缩短与相应的X―H伸缩振动频率红移或蓝移存在较强的相关性,这也是氢键光谱检测和研究的基础.但是,最近的理论研究却推翻了这一观点,认为X―H键长变化和相应的X―H伸缩振动频率移动在有些氢键体系中并不存在相关性(McDowell,S.A.C.; Buckingham,A.D.J.Am.Chem.Soc.2005,127,15515.).本文中,我们采用更为可信的计算方法,对这一问题进行再研究.结果表明是错误的计算方法导致了McDowell和Buckingham得出错误的结论.在McDowell和Buckingham所研究的氢键体系中,X―H键长变化和相应的X―H伸缩振动频率移动仍存在较强的相关性.
氢键复合物;相关;键长变化;振动频率移动;密度泛函理论
1 Introduction
Hydrogen bond is one of the most important noncovalent interactions,which plays key roles in crystal engineering,supramolecular assembly,materials science,and biological system.1,2Infrared and Raman spectroscopies have played important roles in the detection of the hydrogen bonds:conventional X―H··· Y hydrogen bond is characterized by weakening of the X―H bond which causes elongation of this bond and a red shift of the X―H stretching frequency,and the blue-shifting X―H··· Y hydrogen bond is characterized by strengthening of the X―H bond which causes contraction of this bond and a blue shift of the X―H stretching frequency.3Obviously,the basis of the spectroscopic detection of the hydrogen bonds is that frequency shifts correlate very well with the bond-length changes and other characteristics of the hydrogen bond.3,4However,based on the ab initio computational results for a series of linear isoelectronic complexes of HCl with the proton acceptors N2,CO, or BF,McDowell and Buckingham5recently claimed that the widely believed correlation between the bond-length change and the frequency shift in the hydrogen-bonded complexes is unreliable.Such a conclusion is unusual,as it overturns the basis of the spectroscopic study of the hydrogen bond.We know now that the calculations of the bond-length change and the frequency shift are highly method-dependent.6Hence,it is reasonable to suppose that this conclusion may be a result of computational artifacts.In recent years,great progress has been made in the development of new quantum chemical methods.7-9It is the time to revisit this important issue by using more reliable computational methods.
The systems selected in the present study are the linear hydrogen-bonded complexes formed by HCl with N2,CO,BF,or CO2(Fig.1).All the complexes considered by McDowell and Buckingham are included here.For the halogen-bonded complexes,we found that,besides the computational method,the coupling between the X―Hal(Hal=Cl,Br,or I)stretching vibration and other vibrations and the anharmonicity of the X―Hal stretching vibration also have great influence on the bond-length change-frequency shift correlation.10For the hydrogen-bonded complexes,the effect of the coupling between the X―H stretching vibration and other vibrations on the bond-length change-frequency shift correlation can be neglected because of the small mass of the H atom.On the other hand, the study by Sándorfy11indicated that the effect of the anharmonicity of the X―H stretching vibration on the bond-length change-frequency shift correlation is also negligible in most cases.So we did not consider the effects of the vibrational coupling and the anharmonicity on the bond-length change-frequency shift correlation in this work.

Fig.1 Linear complexes considered in this workThe red dot lines represent the hydrogen bonds.
2 Computational details
Electronic structure calculations were carried out using the Gaussian 09 programs.12All the structures were fully optimized with“verytight”convergence criteria and characterized by frequency computations and wave function stability checks.Four newly developed density functionals,M05-2X,13,14M06-2X,14,15B2PLYP-D,16,17and mPW2PLYP-D,17,18were employed to investigate the correlation between bond-length change and vibrational frequency shift in hydrogen-bonded complexes.In order to examine the reliability of these density functionals,MP2, QCISD,and CCSD(T)calculations were also carried out.Both Popleʹs basis sets and Dunningʹs correlation-consistent series of basis sets with diffuse functions were used.The benchmark interaction energies in this work were calculated using the estimated CCSD(T)/CBS procedure,which can be found elsewhere.19An“ultrafine”integration grid(99 radial,590 angular points)was used for all the density functional theory(DFT)calculations to avoid the possible integration grid errors.The binding energies of the complexes were calculated using the supermolecule method.All binding energies are corrected for basis set superposition error(BSSE)using the counterpoise method of Boys and Bernardi.20
In the present study,the value of the bond-length change is given as the difference of the bond length between the complex and the monomer,so that a negative value of the bond-length change refers to a bond contraction and a positive value of the bond-length change indicates a bond elongation.Similarly,a negative value of the frequency shift refers to a red shift and a positive value of the frequency shift means a blue shift.
3 Results and discussion
3.1 Reliability of the M05-2X,M06-2X,B2PLYP-D, and mPW2PLYP-D density functionals

Fig.2 Potential energy curves for the linear complexCl―H···O≡C at different levels of theory All results reflect counterpoise correction.

Many newly developed density functionals have been prov-en to provide improved performance over conventional MP2 method for noncovalent interactions.7-9In this work,we used four popular density functionals M05-2X,M06-2X,B2PLYP-D,and mPW2PLYP-D for the calculations.The hybrid meta density functionals M05-2X and M06-2X are from Truhlarʹs group and the double hybrid functionals B2PLYP-D and mPW2PLYP-D are from Grimmeʹs group.13-18In order to examine the reliability of these density functionals for the hydrogen-bonded complexes considered in the present study,we selected the linear complex Cl―H···O≡C as a model complex and calculated its potential energy curve at the M05-2X/ 6-311++G(3d,3p),M06-2X/6-311++G(3d,3p),B2PLYP-D/ 6-311++G(3d,3p),mPW2PLYP-D/6-311++G(3d,3p),MP2/ 6-311++G(2d,2p),QCISD/6-311++G(2d,2p),QCISD/6-311++ G(3df,3pd),and CCSD(T)/CBS levels of theory,respectively. The highly accurate CCSD(T)/CBS potential energy curve was used as our benchmark.As clearly shown in Fig.2,the MP2/ 6-311++G(2d,2p)and QCISD/6-311++G(2d,2p)calculations underestimate the binding energies and the mPW2PLYP-D/ 6-311++G(3d,3p)calculation tends to be overbound relative to the CCSD(T)/CBS benchmark.More important is the slope of the potential energy curve.It,in fact,determines how accurate the bond length and the vibrational frequency will be.If the two potential energy curves generated at the QCISD/6-311++ G(2d,2p)and QCISD/6-311++G(3df,3pd)levels of theory are removed from Fig.2,we can see that all the potential energy curves are almost in parallel with each other.Evidently,the slopes of the two potential energy curves generated at the QCISD/6-311++G(2d,2p)and QCISD/6-311++G(3df,3pd)levels of theory are inaccurate relative to the CCSD(T)/CBS benchmark,and furthermore they can not produce the correct results of the bond-length change and the frequency shift.Let us add here that McDowell and Buckinghamʹs conclusions are based on the QCISD/6-311++G(2d,2p)and QCISD/6-311++ G(3df,3pd)calculations.5In comparison with the CCSD(T)/ CBS benchmark,the similar slopes of the potential energy curves generated at the M05-2X/6-311++G(3d,3p),M06-2X/ 6-311++G(3d,3p),B2PLYP-D/6-311++G(3d,3p),and mPW2PLYP-D/6-311++G(3d,3p)levels of theory confirm the reliability of the density functionals M05-2X,M06-2X,B2PLYP-D,and mPW2PLYP-D for the study of the bond-length change-frequency shift correlation of the hydrogen-bonded complexes considered in the present study.
3.2 Selection of the basis set
The linear hydrogen-bonded complexes selected in the present study is very small,so it is possible to use very large basis set to eliminate the effect of the basis set superposition error and the basis set incompleteness error.However,considering the possible extension to large hydrogen-bonded complexes,it is meaningful to study the effect of the basis set on the bond length and the vibrational frequency here.
Table 1 lists the optimized geometries,Cl―H stretching frequencies and intensities,dipole moments,and BSSE-corrected binding energies of the studied complexes calculated at the M05-2X/6-311++G(d,p),M05-2X/6-311++G(3d,3p),and M05-2X/6-311++G(3df,3pd)levels of theory,respectively.Compar-ing the values in Table 1,it is found that the results calculated at the M05-2X/6-311++G(d,p)level of theory are obviously different from those calculated at the M05-2X/6-311++G(3d, 3p)and M05-2X/6-311++G(3df,3pd)levels of theory,for instance,the values of the Cl―H bond length calculated at the M05-2X/6-311++G(d,p)level of theory are all larger than the corresponding ones calculated at the M05-2X/6-311++G(3d, 3p)and M05-2X/6-311++G(3df,3pd)levels of theory,whereas the values of the Cl―H stretching frequency calculated at the M05-2X/6-311++G(d,p)level of theory are larger or smaller than the corresponding ones calculated at the M05-2X/6-311++ G(3d,3p)and M05-2X/6-311++G(3df,3pd)levels of theory;at the same time,it is also noticed that the results calculated at the M05-2X/6-311++G(3d,3p)level of theory are almost the same as those calculated at the M05-2X/6-311++G(3df,3pd) level of theory.These results indicate that the basis set 6-311++ G(3d,3p)yields converged results.In the following studies,we will use the basis set 6-311++G(3d,3p)for the calculations.

Table 1 Optimized geometries(r and R,in nm),Cl―H stretching frequencies(ν,in cm-1)and intensities(I,in km·mol-1),dipole moments(μ,in C·m),and BSSE-corrected binding energies(ΔECP,in kJ·mol-1)of the studied complexes calculated using M05-2X functional with basis sets 6-311++G(d,p),6-311++G(3d,3p),and 6-311++G(3df,3pd)
3.3 Cl―H bond-length change versus the Cl―H stretching frequency shift
The values of Cl―H bond-length change and the corresponding Cl―H stretching frequency shift upon the hydrogen-bonded complex formation are summarized in Table 2 and plots of the Cl―H bond-length change versus the corresponding Cl―H vibrational frequency shift are shown in Fig.3.

Fig.3 Correlation of Cl―H bond-length change with Cl―Hstretching frequency shift in hydrogen-bonded complexes
McDowell and Buckingham found,at their highest level of theory(QCISD/6-311++G(3df,3pd)),that blue shifts were obtained for Cl―H···F―B and Cl―H···O≡C,while red shifts were obtained for the other hydrogen-bonded complexes.5Surprisingly,a blue shift accompanied by a Cl―H bond elongation was also predicted for Cl―H···O≡C.5Here in Table 2, our results show that blue shift is obtained only for Cl―H···F―B,while Cl―H···O≡C and other complexes are all bound by the red shifting hydrogen bonds.At the same time,it can be seen from Table 2 that all the data are very normal,that is to say,the red shift is connected with the bond elongation and the blue shift is connected with the bond contraction.As mentioned above,it is the computational artifact that leads to the different results.Note that McDowell and Buckingham also calculated Cl―H···O≡C at the MP2/6-311++G(2d,2p)level of theory.Their MP2 results are in agreement with our results.This can be explained by the correct slope of the potential energy curve of MP2 calculations,as mentioned above.
The correlation between the bond-length change and the frequency shift for the Cl―H bond can be seen in Fig.3.Plots of the Cl―H bond-length change versus the corresponding vibrational frequency shift of the Cl―H stretch all give straight lines.The coefficient of determination of these linear fits(R2) is about 0.995 at the M05-2X/6-311++G(3d,3p)and M06-2X/ 6-311++G(3d,3p)levels of theory and about 0.999 at the B2PLYP-D/6-311++G(3d,3p)and mPW2PLYP-D/6-311++G(3d,3p) levels of theory.These results show that the correlation between the Cl―H bond-length change and the corresponding frequency shift is excellent in the hydrogen-bonded complexesstudied.
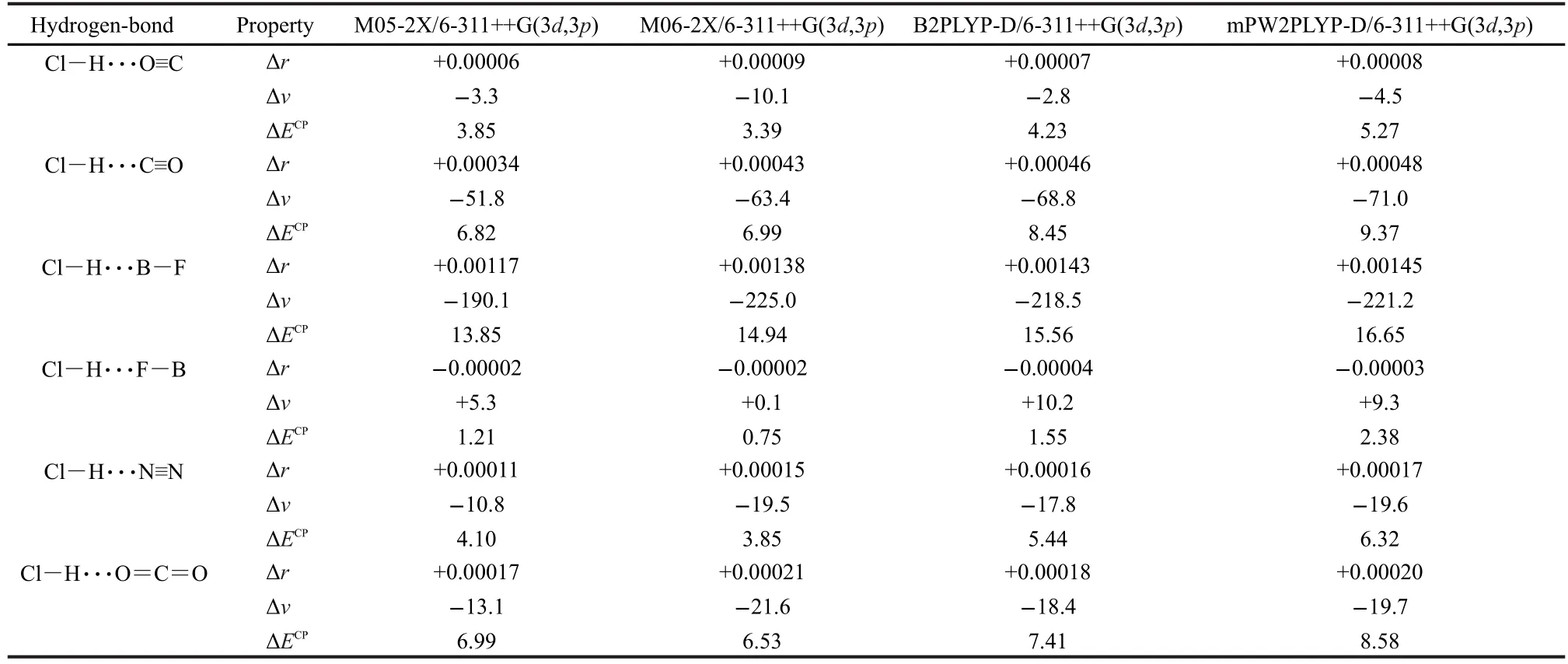
Table 2 Cl―H bond-length changes(Δr,in nm)and the corresponding Cl―H stretching frequency shifts(Δν,in cm-1)upon the hydrogen-bonded complexes formation,and the BSSE-corrected binding energies(ΔECP,in kJ·mol-1)of the studied complexes at various levels of theory
4 Conclusions
The Cl―H bond-length change and the corresponding vibrational frequency shift of the Cl―H stretch upon the hydrogen bond formation have been determined using high level ab initio and density functional theory computations.It is found that the bond-length change-frequency shift correlation is highly method-dependent.The computational artifacts can lead to false conclusions.Compared with the CCSD(T)benchmark, M05-2X,M06-2X,B2PLYP-D,and mPW2PLYP-D give reasonable results,whereas the wave function theory-based QCISD performs poorly.Employing more reliable M05-2X, M06-2X,B2PLYP-D,and mPW2PLYP-D calculations,we found that plots of the Cl―H bond-length change versus the corresponding vibrational frequency shift of the Cl―H stretch all give straight lines and the coefficients of determination of the fits are close to 1 in all cases,showing that the correlation between the Cl―H bond-length change and the corresponding frequency shift is excellent in the hydrogen-bonded complexes studied.The correlation also indicates that the red shift is connected with the bond elongation,whereas the blue shift is connected with the bond contraction,and vice versa.We believe that,in addition to the Cl―H bond studied here,the results for other X―H bonds should be the same.
The present study focuses only on the correlation between bond-length change and vibrational frequency shift of the hydrogen bond in the ground state.It is unclear whether the correlation still remains for the hydrogen bond in the electronically excited state.21Further study is underway in our laboratory.
(1) Desiraju,G.R.Angew.Chem.Int.Edit.2011,50,52.
(2) Arunan,E.;Desiraju,G.R.;Klein,R.A.;Sadlej,J.;Scheiner, S.;Alkorta,I.;Clary,D.C.;Crabtree,R.H.;Dannenberg,J.J.; Hobza,P.;Kjaergaard,H.G.;Legon,A.C.;Mennucci,B.; Nesbitt,D.J.Pure Appl.Chem.2011,83,1619.
(3) Hobza,P.;Havlas,Z.Chem.Rev.2000,100,4253.
(4) Li,X.;Liu,L.;Schlegel,H.B.J.Am.Chem.Soc.2002,124, 9639.
(5)McDowell,S.A.C.;Buckingham,A.D.J.Am.Chem.Soc. 2005,127,15515.
(6) Lu,P.;Lin,G.Q.;Li,J.C.J.Mol.Struct.-Theochem 2005,723, 95.
(7) Sun,T.;Wang,Y.B.Acta Phys.-Chim.Sin.2011,27,2553. [孙 涛,王一波.物理化学学报,2011,27,2553.]
(8) Schwabe,T.;Grimme,S.Accounts Chem.Res.2008,41,569.
(9) Sherrill,C.D.J.Chem.Phys.2010,132,110902.
(10) Wang,W.;Zhang,Y.;Ji,B.;Tian,A.J.Chem.Phys.2011,134, 224303.
(11) Sándorfy,C.J.Mol.Struct.2006,790,50.
(12) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 09, Revision C.01;Gaussian Inc.:Wallingford CT,2010.
(13) Zhao,Y.;Truhlar,D.G.J.Chem.Theory Comput.2006,2,1009.
(14) Zhao,Y.;Truhlar,D.G.Accounts Chem.Res.2008,41,157.
(15) Zhao,Y.;Truhlar,D.G.Theor.Chem.Acc.2008,120,215.
(16) Grimme,S.J.Chem.Phys.2006,124,034108.
(17) Schwabe,T.;Grimme,S.Phys.Chem.Chem.Phys.2007,9, 3397.
(18) Schwabe,T.;Grimme,S.Phys.Chem.Chem.Phys.2006,8, 4398.
(19) Pitoňák,M.;Janowski,T.;Neogrády,P.;Pulay,P.;Hobza,P. J.Chem.Theory Comput.2009,5,1761.
(20) Boys,S.F.;Bernardi,F.Mol.Phys.1970,19,553.
(21) Zhao,G.J.;Han,K.L.Accounts Chem.Res.doi:10.1021/ ar200135h.
November 4,2011;Revised:December 27,2011;Published on Web:December 30,2011.
Correlation between Bond-Length Change and Vibrational Frequency Shift in Hydrogen-Bonded Complexes Revisited
ZHANG Yu MA Ning WANG Wei-Zhou*
(College of Chemistry and Chemical Engineering,Luoyang Normal University,Luoyang 471022,Henan Province,P.R.China)
The correlation between the X―H bond-length change and the corresponding X―H stretching frequency shift upon X―H···Y(Y is an electron donor)hydrogen bond formation is the basis for the spectroscopic detection and investigation of the hydrogen bond.However,this view has been questioned in a recent report,suggesting that the widely accepted correlation between the bond-length change and the frequency shift in hydrogen-bonded complexes is unreliable(McDowell,S.A.C.;Buckingham,A.D.J.Am. Chem.Soc.2005,127,15515.).In this work,several robust computational methods have been used to investigate this issue.The results clearly show that a computational artifact leads to the conclusion incorrectly reported by McDowell and Buckingham and that the correlation between the X―H bond-length change and the corresponding X―H stretching frequency shift is still very good in the hydrogen-bonded complexes studied.
Hydrogen-bonded complex;Correlation;Bond-length change;Vibrational frequency shift; Density functional theory
10.3866/PKU.WHXB201112303
O641
∗Corresponding author.Email:wzwanglab@yahoo.com;Tel:+86-379-65515113.
The project was supported by the National Natural Science Foundation of China(21173113),Aid Project for the Leading Young Teachers in Henan Provincial Institutions of Higher Education of China(2010GGJS-166),and Natural Science Foundation of Henan Educational Committee,China (2010A150017,2011B150024).
国家自然科学基金(21173113),河南省高等学校青年骨干教师资助计划项目(2010GGJS-166)和河南省教育厅自然科学研究计划项目(2010A150017,2011B150024)资助
猜你喜欢
中学生理科应试(2024年1期)2024-05-18 13:02:52
无机化学学报(2024年1期)2024-01-20 03:55:50
化工职业技术教育(2021年5期)2021-11-09 03:10:40
化工职业技术教育(2021年1期)2021-03-15 06:57:44
青岛大学学报(工程技术版)(2019年2期)2019-09-10 07:22:44
化工学报(2016年3期)2016-03-14 08:37:00
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
中学化学(2015年8期)2015-12-29 07:32:44
原子与分子物理学报(2014年3期)2014-02-28 22:18:23
