
放射敏感性启动子调控CD基因/5-FC自杀系统慢病毒载体的构建及125I诱导表达的研究
2012-11-12 07:09张春丽王荣福
同位素 2012年3期
李 玲,张春丽,闫 平,殷 雷,康 磊,赵 倩,王荣福
(北京大学 第一医院 核医学科,北京 100034)
放射敏感性基因“启动子”序列可在电离辐射作用下诱导下游基因表达,将放射敏感性启动子序列连接于自杀基因DNA 的上游,构建重组慢病毒载体,包装重组慢病毒颗粒,以重组慢病毒感染细胞,以放射性核素的电离辐射作用作为启动自杀基因转录的开关,在空间和时间上调控自杀基因的表达,通过放射性核素的电离辐射效应联合基因治疗,可在肿瘤组织局部产生协同抗肿瘤细胞作用,从而在提高对肿瘤组织的杀伤作用的同时,减少辐射损伤和化疗药物治疗造成的全身毒副效应[1]。
Marples、Scott[2-3]等的研究发现,上游基因序列含CArG 元件即CC(A/T)6GG 序列,可在电离辐射诱导下成功诱导下游基因表达,与电离辐射可诱导的野生Egr-1基因上游启动子相比,含4个及以上CArG 元件的合成启动子在受到相同剂量辐射时,其下游基因表达明显增加,P<0.05;而含有6~9个CArG 元件时,报告基因表达可增至最高。
胞嘧啶脱氨酶(Cytosine Deaminase,CD)/5-FC(5-Fluorocytosine,5-氟胞嘧 啶)为目 前研究最多的自杀基因/前药系统之一。CD 基因表达产物为胞嘧啶脱氨酶,能将抗真菌药物5-FC转化为细胞毒性药物5-FU(5-Fluorourial,5-氟尿嘧啶),5-FU 在细胞内的代谢物通过插入DNA 或RNA 链,抑制DNA 或蛋白质的合成,从而发挥杀灭细胞作用。同时5-FU 也是一种放疗増敏剂,其増敏机理可能为减少细胞核酸形成、抑制细胞修复、减少S期的抗放疗细胞。且有文献[4]报道,在CD/5-FC 基因治疗中,仅2%~10%的肿瘤细胞表达CD 基因,就可明显抑制肿瘤生长,称之为基因治疗的“旁观者效应”。
慢病毒(Lentivirus)载体是近年来以HIV-1(人类免疫缺陷Ⅰ型病毒)为基础发展起来的基因治疗载体,对分裂细胞和非分裂细胞均具有感染能力,可将外源基因有效地整合到宿主细胞的染色体上,从而达到持久性表达,因其去除了毒性基因,故安全性高,因而在基因治疗方面得到了广泛的应用[5]。
本工作拟构建含放射性启动子E8、自杀基因CD 基因及报告基因GFP(Green Fluorescent Protein,绿色荧光蛋白)基因的重组慢病毒载体,包装成重组慢病毒颗粒E8-codA-GFP LV,感染EJ细胞,以放射性核素125I诱导合成的放射敏感性启动子E8(含8个CArG 元件的DNA序列)启动CD 基因及GFP 基因表达,将5-FC转化成5-FU,并表达绿色荧光,通过细胞体系中5-FU 的检测及对绿色荧光表达的观察,以验证构建的放射敏感性启动子E8 调控CD 基因/5-FC自杀系统慢病毒载体具有电离辐射调控作用。
1 实验材料
1.1 主要试剂与材料
慢病毒表达质粒pGC-FU-GFP 载体、慢病毒结构蛋白质粒pHelper 1.0 载体及慢病毒包膜蛋白质粒pHelper 2.0载体(含VSVG 元件):GeneChem公司产品;含CD基因的pCD2质粒:ATCC公司产品;293T 细胞:上海吉凯基因技术有限公司提供;LipofetamineTM2000:Invitrogen公司产品;膀胱癌EJ细胞:北京大学医学部病理教研室提供;Plasmid 抽提试剂盒:Promega公司产品;TRIZOL 试剂盒:Invitrogen公司产品。AgeⅠ酶、PacⅠ/Bam H Ⅰ酶:NEB公司产品。
5-FC、5-FU:Sigma公司产品;RPMI1640培养基、DMEM培养基及FBS(Fetal Blood Serum,胎牛血清):Hyclone公司产品;ENi.s、Polybrene:上海吉凯基因技术有限公司提供;Na125I溶液(2.22TBq/L,放化纯度>99%):原子高科股份有限公司产品。
1.2 主要仪器与设备
PCR 仪:Applied Biosystems公司 产 品:稳压DNA 电泳仪:BioRad公司产品;高速离心机:日立公司产品;凝胶成像仪:天能公司产品;Class-VP SCL-10AVP HPLC分析型:岛津公司产品;VenusilMPC-18制备色谱柱(10mm×250mm,5μm):Alltech 公司产品;荧光显微镜:奥林巴斯公司产品;COM2 培养箱:Thermo公司产品。
2 实验方法
2.1 重组慢病毒载体构建与重组慢病毒包装
2.1.1 CD 基因获取、扩增及载体pGC-FU-codA-GFP制备
根据已知GeneBank 中CD 基因序列设计合成引物,分别在引物上下游加入AgeⅠ酶切位点,由上海捷瑞生物工程有限公司合成。其引物序列:CD基因序列-Age Ⅰ-F 为5’GAGGATCCCCGGGTACCGGTCGCCACCATGTC GAATAACGCTTTACAAAC3’;CD基因序列-AgeⅠ-R为5’TCACCATGGTGGCGACCGGACGTTTGTAATCGATGGCTTC3’。以含CD 基因序列 的质粒pCD2为模板,PCR扩增,获取CD基因cDNA。PCR(Polymerase Chain Reaction,聚合酶链式反应)反应条件:94℃变性5 min;设置参数:94℃变性30s、55℃退火30s、72℃延伸2min,循环30次;最后72℃延伸10min。电泳鉴定PCR 产物。
AgeⅠ酶切消化pGC-FU 载体,使含GFP基因的慢病毒载体线性化后,加入纯化的PCR产物,使之通过交换进入线性化慢病毒载体。其反应体系包括2.5μL 双蒸水、2μL n-Fusion交换酶缓冲液、5μL线性化载体DNA、10μL纯化的PCR 产物、0.5μL In-Fusion交换酶,总体积20μL。于25℃下反应30min,再加热至42℃反应15min,制得交换液,转化经CaCl2处理过的大肠杆菌感受态细胞DH5α,PCR 鉴定细菌阳性克隆,接种阳性转化子,抽提质粒,获得载体pGC-FU-codA-GFP,并由上海美季生物技术有限公司测序验证。
2.1.2 放射性启动子E8 合成及慢病毒载体pGC-FU-E8-codA-GFP制备
化学合成含8个CArG 元件的启动子,两端为PacⅠ/BamHⅠ酶切位点,其序列为5’TTAATTAACCGCGGCCTTATTTGGCCT TATTTGGCCTTATTTGGCCTTATTTGGCCTTATTTGGCCTTATTTGGCCTTATTTG GCCTTATTTGGCCGCGGGGATCC3’,由Invitrogen公司合成。Pa cⅠ/Bam H Ⅰ酶切化学合成的含有目的基因的质粒。Pac Ⅰ/BamHⅠ酶切消化pGC-FU-codA-GFP使之线性化后,加入酶切的启动子DNA 片段,制备pGC-FUE8-codA-GFP。其反应体系包括:13.5μL ddH2O、2μL T4DNA 连接酶缓冲液、2μL 50%PEG4000、1μL酶切载体DNA、0.5μL 酶切纯化的PCR 产物、1μL T4DNA 连接酶,总体积20μL。于22℃下连接3h,之后转化经CaCl2处理过的大肠杆菌感受态细胞DH5α,PCR 鉴定细菌阳性克隆,接种阳性转化子,抽提质粒,获得载体pGC-FU-E8-codA-GFP,由上海美季生物技术有限公司测序验证。
2.1.3 重组慢病毒包装
将重组慢病毒载体pGC-FU-E8-codAGFP、空载慢病毒载体pGC-FU 载体以及慢病毒包装辅助质粒pHelper 1.0载体、pHelper 2.0载体分别进行高纯度无内毒素抽提,按Lipofectamine2000试剂使用说明,分别用重组载体、空载体与辅助质粒共转染293T 细胞,培养8h后去除转染液,更换为原培养液。于转染后48h收集细胞上清液,4℃、4 000g离心10min,除去细胞碎片,以0.45μm 滤器过滤上清液,浓缩后获得纯化含目的基因的重组慢病毒浓缩液及仅含GFP基因的空载慢病毒浓缩液,即E8-codA-GFP LV 及GC-FU-GFP LV,各自分装后存于-80℃中备用。
2.2 慢病毒滴度测定
用逐孔稀释法测定慢病毒滴度:将处于对数生长期的293T 细胞按每孔1×105个细胞接种于24 孔板中。取病毒浓缩液用ENi.s(Enhanced Infection Solution,感染增强液)按10-1、10-2、10-3、10-4、10-5、10-6进行梯度稀释后感染细胞,37℃、5%CO2培养箱中培养48h,换液。4d后,观察空载慢病毒组荧光表达情况,并计算空载慢病毒滴度。重组目的基因慢病毒组用Realtime 定量PCR 法测定E8-codA-GFP LV 滴度:抽提E8-codA-GFP LV 感染组细胞总RNA后,逆转录成cDNA,用Realtime 定 量PCR 法检测GFP 基因表达,以ACTIN 作为内参照。
2.3 慢病毒感染EJ细胞
人膀胱癌EJ细胞培养:用含10%FBS 的RPMI 1640培养基将EJ细胞培养于37℃、5%CO2培养箱中。将生长状态良好的EJ细胞按每孔3×103个接种于96孔板上,至细胞融合率达50%时,用ENi.s稀释空载病毒原液,以MOI(Multiplicity of Infection,感染复数)值为1、10、20、40、60、80、100感染EJ细胞,荧光显微镜下观察绿色荧光,确定EJ细胞最佳MOI值。目的慢病毒E8-codA-GFP LV 用ENi.s稀释,以最佳MOI值感染EJ细胞。
2.4 荧光显微镜观察绿色荧光
E8-codA-GFP LV 感染EJ细胞后48h,吸弃原培养液,分别加入0、18.5、37.0、55.5、74.0kBq用培养液稀释过的Na125I,补足培养液至终体积为100μL。继续于37℃、5%CO2培养箱中培养,48h后置于荧光显微镜下观察。
2.5 HPLC检测细胞上清液中的5-FU
E8-codA-GFP LV 感染EJ细胞后48h,吸弃原培养液,将感染细胞分为11 组,1~5 组分别加入0、37、74、111、148kBq Na125I,每组同时加入40μL 5-FC(1g/L),6~10组每组分别加入0、37、74、111、148kBq Na125I,第11组仅加入40μL 5-FU(1 mg/mL),各孔补足培养液至总体积100μL,继续培养于37℃、5%CO2培养箱中。48h 后,吸取上清液,以乙腈和水为展开剂,用HPLC法分离并观察细胞上清液中5-FU紫外峰。
3 结果与讨论
3.1 慢病毒载体构建与DNA 测序
以pCD2质粒为模板,PCR 扩增CD 基因序列,结果示于图1。由图1 可知,扩增产物大小约1 327bp。
PacⅠ/Bam H Ⅱ酶切凝胶电泳图示于图2。由图2可知,PacⅠ/Bam H Ⅰ酶切化学合成的含有启动子序列的质粒,其酶切片段大小96bp。
连接产物转化感受态大肠杆菌DH5α 后,PCR 鉴定细菌阳性克隆。阳性转化子进行DNA 测序比对,结果显示,重组慢病毒载体pGC-FU-E8-codA-GFP质粒中含有合成正确的8个CC(A/T)6GG 放射敏感性启动子序列及目的基因codA,与Gennebank中X63656.1序列基本一致(CD 基因为原核生物大肠杆菌E.coli中获得,其起始密码子为GUG,而真核生物中起始密码子为AUG,故第一位碱基由基因收录库中的G 突变为A)[6]。
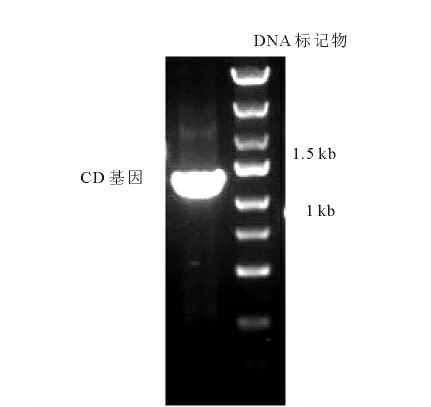
图1 CD基因PCR 产物凝胶电泳图

图2 E8启动子酶切凝胶电泳图
3.2 慢病毒滴度测定结果
空载慢病毒载体及重组目的慢病毒载体分别与慢病毒包装辅助质粒共转染293T 细胞,成功获得空载慢病毒及重组目的慢病毒颗粒。经逐孔稀释法转染293T 细胞后,通过观察感染细胞各组绿色荧光表达,计算得到空载慢病毒液滴度为2×109TU/mL。通过Realtime定量PCR法检测,计算得到重组目的慢病毒液滴度为2×108TU/mL。
3.3 慢病毒转染EJ细胞观察绿色荧光表达
空载慢病毒以不同MOI值感染EJ细胞,荧光显微镜观察绿色荧光表达,结果显示,MOI=60(即每孔中慢病毒数/细胞数=60)时荧光强度增至最高,其显微图像示于图3。重组目的慢病毒感染EJ细胞,加入不同剂量125I后绿色荧光表达结果示于图4。由图4可见,125I用量对重组目的慢病毒感染EJ细胞的绿色荧光表达结果影响明显,未加125I时,基本无荧光表达(图4a);随着125I用量的增加,荧光表达也在逐渐增加,37.0Bq时即有明显表达(图4c),74.0kBq组(图4e)荧光表达最明显。可见,合成的放射敏感性启动子可在低剂量放射性核素125I的电离辐射作用下诱导下游基因GFP基因表达。
3.4 HPLC结果
对2.5节中1~5组的细胞上清液及20μL 5-FC、5-FU 标准对照液进行HPLC 分析,结果显示,于254nm 可观察到紫外吸收峰,保留时间5min左右可观察到5-FC峰,6min左右可观察到5-FU 峰。加入不同剂量125I对400mg/L 5-FC组其HPLC 谱示于图5。图5 中,第一个峰为5-FC,第二个峰为5-FU。由图5 可知,加入148kBq125I时,5-FU 紫外峰最为明显(图5d)。以上结果说明,在放射性核素125I诱导作用下,感染了目的慢病毒的EJ细胞能够将5-FC转化成原本不存在的5-FU,且随着125I剂量的增加,5-FC转化生成5-FU 的量有所增加,加入148kBq125I时,5-FU 生成量最多。
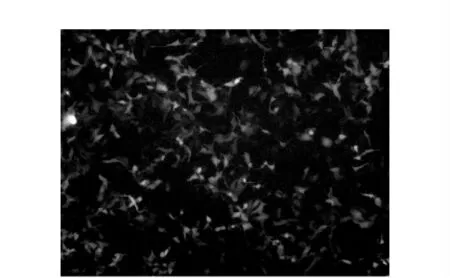
图3 空载慢病毒感染EJ细胞后的绿色荧光表达

图5 不同剂量125I诱导E8-codA-GFP LV感染EJ细胞转化5-FC为5-FU HPLC谱图
4 结论
1)本实验构建的重组慢病毒载体pGC-FUE8-codA-GFP序列正确,并可成功包装出滴度较高的慢病毒颗粒E8-codA-GFP LV。
2)重组慢病毒E8-codA-GFP LV 能够感染人膀胱癌EJ细胞,实验中设计合成的含8 个CArG 元件的放射敏感性启动子序列在可在较低剂量放射性核素的电离辐射作用下诱导下游基因CD 基因及GFP基因的表达,成功将5-FC转化成5-FU,并表达荧光蛋白。
以上结果提示,低能量的放射性核素125I在较低剂量下(37kBq)即可诱导放射敏感性启动子E8调控下游基因表达,激活下游基因CD,将5-FC转化成细胞毒性药物5-FU。由此可见,若将发射低能量射线的放射性核素如125I连接于可与肿瘤细胞和组织特异性结合的单克隆抗体或者小分子上,在体内靶向作用于转入外源放射性增敏基因(如放射敏感性启动子E8)及治疗基因的肿瘤细胞,诱导治疗基因表达,将无毒性前体药物在肿瘤局部转化成细胞毒性药物,即可发挥放射免疫治疗和基因治疗的双重作用,且可以通过自杀系统产生的“旁观者效应”进一步扩大其作用范围,对未转入外源基因的肿瘤细胞发挥作用,以达到降低细胞毒性药物全身毒副作用时,在局部充分发挥其抗肿瘤作用。
但本实验中尚存在诸多不足之处,如慢病毒感染细胞中导入的外源基因所需的表达时间较长。实验中125I诱导下游基因的表达尚需要进一步探索更为合适的实验条件,以观察到更为全面的表达效果,还需要进一步研究125I剂量对基因表达的影响,为下一步的实验提供更好的条件。
[1]王荣福,张春丽.电离辐射诱导启动子Egr-1调控的基因放射治疗[J].肿瘤学杂志,2010,16(3):238-241.
[2]Marples B,Scott SD,Hendry JH,et al.Development of synthetic promoters for radiation-mediated gene therapy[J].Gene Ther,2000,7(6):511-517.
[3]Scott SD,Joiner MC,Marples B.Optimizing radiation-responsive gene promoters for radiogenetic cancer therapy[J].Gene Ther,2002,9(20):1 396-1 402.
[4]Huber BE,Austin EA,Richards CA,et al.Metabolism of 5-fluorocytosine to 5-fluorouracil in human colorectal tumor cells transduced with the cytosine deaminase gene:Significant antitumor effects when only a small percentage of tumor cells express cytosine deaminase[J].Proc Natl Acad Sci USA,1994,91(17):8 302-8 306.
[5]Dropuli B.Lentiviral vectors:Their molecular design,safety,and use in laboratory and preclinical research[J].Hum Gene Ther,2011,22(6):649-657.
[6]Danielsen S,Kilstrup M,Barilla K,et al.Characterization of the Escherichia coli codBA operon encoding cytosine permease and cytosine deaminase[J].Mol Microbiol,1992,6(10):1 335-1 344.
猜你喜欢
华人时刊(2022年9期)2022-09-06
空间科学学报(2021年1期)2021-05-22
江西农业学报(2021年4期)2021-04-20
水生生物学报(2021年1期)2021-02-04
华人时刊(2020年15期)2020-12-14
畜牧与饲料科学(2018年11期)2018-12-10
中国核电(2018年3期)2018-10-10
中国医学装备(2018年7期)2018-07-12
科学家(2017年19期)2017-10-27
中国当代医药(2015年9期)2015-03-01
