
白细胞介素-1β在病理性疼痛大鼠脊髓LTP中的作用及机制*
2012-11-06 04:03黄阳亮许继德
中国病理生理杂志 2012年3期
钟 祎, 黄阳亮, 许继德
(1广州医学院生理教研室,广东 广州 510182 ;2中山大学附属第一医院脊柱外科,广东 广州 510700)
1000-4718(2012)03-0541-05
2011-09-20
2012-01-05
广州医学院博士启动基金资助项目(No.2010C12)
△通讯作者 Tel:020-81340199; E-mail:victoria0720@126.com
白细胞介素-1β在病理性疼痛大鼠脊髓LTP中的作用及机制*
钟 祎1△, 黄阳亮2, 许继德1
(1广州医学院生理教研室,广东 广州 510182 ;2中山大学附属第一医院脊柱外科,广东 广州 510700)
目的探讨白细胞介素-1β (interleukin-1β, IL-1β)在神经病理性疼痛大鼠脊髓背角C纤维诱发电位长时程增强(long-term potentiation, LTP)中的作用及其机制。方法用坐骨神经部分损伤(spared nerve injury, SNI)和腰5前根切断(lumbar 5 ventral root transection, L5 VRT)方法复制大鼠病理性疼痛模型,观察外源性IL-1β对正常大鼠及病理性疼痛模型大鼠脊髓背角C纤维诱发电位的影响,并且检测p38 MAPK (p38 mitogen-activated protein kinase)和NF-κB (nuclear factor-kappa B)信号通路在其中的作用。结果500 μg/L IL-1β对正常大鼠C纤维介导的基本突触传递和高频刺激诱导的LTP都没有影响,而5 μg/L的IL-1β可以在神经病理性疼痛模型的大鼠上诱导出LTP。预先用p38 MAPK或NF-κB的抑制剂(SB203580或PDTC)可以完全阻断IL-1β诱导的LTP。结论外源性IL-1β可诱导神经病理性疼痛大鼠脊髓背角C纤维诱发电位的LTP。p38 MAPK和NF-κB信号通路可能参与这一过程。
白细胞介素-1; 长时程增强; p38 MAPK; NF-κB
突触传递效率的长时程增强(long-term potentiation, LTP)现象最早是在海马发现的,是现在公认的研究学习和记忆的突触模型[1]。在中枢神经系统的其它部位比如脊髓也发现电刺激传入C纤维以及末梢神经损伤可以诱导脊髓背角LTP[2-3]。由于传入C纤维传递痛觉信息并与脊髓浅层神经元形成突触联系,脊髓LTP被认为是一种痛觉记忆,是目前研究病理性疼痛的突触模型[4]。
大量文献表明,海马LTP和脊髓LTP有着许多相同的机制。蛋白激酶C (protein kinase C, PKC)、蛋白激酶A (cAMP-dependent protein kinase A, PKA)和钙离子/钙调蛋白依赖性激酶II (calcium/calmodulin-dependent protein kinases II, CaMKII)在海马LTP和脊髓LTP的作用方式相同[5]。激活多巴胺D1/D5受体或者原肌球蛋白相关受体B(tropomyosin-related kinase B, TrkB)受体可以诱导海马和脊髓背角LTP[6-7]。很显然,以这些分子为靶点的药物可以治疗病理性疼痛但同样会带来严重的并发症——记忆功能受损。因此选择性地抑制脊髓LTP非常重要。
致炎细胞因子比如肿瘤坏死因子 α (tumor necrosis factor α, TNF-α)和白细胞介素-1β (interleukin-1β,IL-1β)在突触可塑性中起重要作用。病理浓度下它们可以抑制海马CA1、CA3和齿状回的LTP[8-9]。本研究的目的就是探讨可以引起病理性疼痛的IL-1β对脊髓LTP的影响及其机制。
材 料 和 方 法
1材料
1.1动物 成年雄性Sprague-Dawley (SD)大鼠,体重220~280 g,均由广州医学院实验动物中心提供。动物分笼饲养,自由饮食。室温及室内相对湿度分别维持在(24±1)℃和(55±5)%,所有实验步骤均按照有关实验动物的使用原则操作。
1.2主要试剂 重组大鼠IL-1β (Calbiochem), PDTC (NF-kappa B抑制剂,Sigma)首先溶解在0.9%生理盐水中配成储存液,分装储存在-80 ℃冰箱中。使用前用0.9%生理盐水稀释成所需浓度。SB203580 (p38 MAPK 抑制剂,Sigma)和SP600125 (JNK抑制剂,Calbiochem)先用DMSO溶解为50 mmol/L的母液,使用前再用0.9%生理盐水稀释。抑制剂预处时间为60 min,处理后吸出剩余抑制剂,再于脊髓表面加入IL-1β观察抑制剂对IL-1β作用的影响,IL-1β的作用时间一直维持到实验结束。所有药物使用前先在恒温水浴箱中预热到37 ℃再加于脊髓表面。
2方法
2.1疼痛模型制作
2.1.1坐骨神经部分损伤(spared nerve injury, SNI)模型[10]2 %氟烷麻醉大鼠,剔除大鼠左大腿毛发,0.5%氯已定消毒手术部位。剪开大腿外侧皮肤,钝性分离肌肉,暴露坐骨神经及其3个分支:腓肠神经、腓总神经和胫神经。用5.0丝线紧紧结扎腓总神经和胫神经,并从结扎远端剪掉2~4 mm。注意不要损伤腓肠神经,伤口分层缝合, 待动物苏醒后,与术前同条件喂养。
2.1.2腰5前根切断(lumbar 5 ventral root transection, L5 VRT)模型[11]苯巴比妥钠(50 mg/kg, 腹腔注射)麻醉动物,所有操作在左侧脊柱进行。手术过程遵循无菌原则。剔除大鼠下背部的毛发,暴露皮肤。0.5%氯已定消毒手术部位,铺手术巾,使用消毒过的手术器械。从大鼠脊柱旁约腰4~骶2(L4~S2)脊髓节段处纵行划开皮肤,去除L5节段上下的肌肉组织,用咬骨钳移去左侧L6横突后暴露L4~L5脊神经,用玻璃分针小心分离L5脊神经并用3.0医用缝合线牢固结扎并切断,逐层缝合切口,待动物苏醒后,与术前同条件喂养。
2.2行为学测试[12]为了让大鼠熟悉测试环境,消除大鼠心理因素对测试结果的影响,于测试前1周把大鼠放于测试箱内15~20 min,隔天1次, 共3 次。于造模后1 d、3 d、7 d、14 d和21 d进行痛行为学测试。测试箱为透明有机玻璃箱(18 cm×25 cm×18 cm),箱底为金属网制成(网格为0.8 cm×0.8 cm),通过网孔用von Frey hair可以对大鼠足底部皮肤施加机械性刺激。刺激后大鼠出现迅速的撤足或舔足视为阳性反应。仅仅出现了撤足阈值明显降低的模型动物才用做电生理实验。
2.3电生理记录 根据行为测试的结果,取造模后7 d的动物(此时已出现明显的痛觉过敏)进行电生理记录。乌拉坦(1.5 g/kg)腹腔注射麻醉,行气管插管使大鼠自然呼吸;一侧颈动脉插管持续监测大鼠血压(波动于80~120 mmHg);在腰4~腰6(L4~L6)间行椎板切除术,暴露腰膨大。游离左侧坐骨神经,使用双极氯化银电极刺激坐骨神经。除用于记录的脊髓节段(腰膨大部位:L4~L6)外,暴露的神经组织均用石蜡油覆盖。用热反馈式电热毯使大鼠肛温维持在37.0~37.5 ℃。脊髓背角C纤维诱发电位的记录方法如下: 通过氯化银电极电刺激游离的左侧坐骨神经以诱发脊髓场电位,钨丝微电极(电阻0.5~1 MΩ)进行细胞外记录,用电控的微量推进器(Narishige Scientific Instrument Laboratory)将电极深度控制在脊髓表面下100~500 μm。用数/模转换器(ADC-42. PICO)以10 kHz的速度处理和储存数据。以C纤维诱发电位的幅度作为参数。单方波(10~20 V,0.5 ms,1 min)刺激分离的左侧坐骨神经诱发脊髓背角电位,高频刺激(40 V,0.5 ms,100 Hz,持续1 s,间隔10 s,4次)诱导C纤维诱发电位LTP。坐骨神经的刺激点到脊髓背角的记录点约为11 cm。每只动物只做1次实验,每只动物电生理记录总时间约为7 h。实验结束后腹腔注射过量的乌拉坦处死动物。
3统计学处理

结 果
1正常大鼠脊髓表面给予IL-1β不影响高频刺激诱导的脊髓背角LTP
图1B显示,在不同浓度的IL-1β (5 μg/L, 50 μg/L, 500 μg/L; 200 μL)的作用下,高频刺激诱导的LTP水平和生理盐水作用下高频刺激诱导的LTP水平无显著差异(P>0.05)。500 μg/L IL-1β 对C纤维介导的基本突触传递亦无显著影响,见图1。
2神经损伤大鼠脊髓表面给予IL-1β可以诱导脊髓背角LTP
在SNI模型大鼠脊髓表面给予浓度低至5 μg/L (200 μL) IL-1β可以引起脊髓背角LTP。给予IL-1β 30 min后,脊髓背角C纤维诱发的场电位明显增加(143.2±11.8)% (P<0.05),165 min后场电位增加到(250.8 ± 17.5)%(P<0.05),并且在此水平维持到实验结束(n=7)。而IL-1β的溶剂生理盐水对C纤维诱发电位没有显著影响(P>0.05),见图2A。IL-1β (5 μg/L, 200 μL)及溶剂生理盐水对正常大鼠的基础电位没有显著影响(P>0.05),见图2C。为了进一步验证这个结果,我们在L5 VRT模型大鼠上进行了相同的实验,结果与前面相似, 脊髓表面给予IL-1β可以诱导脊髓LTP (P<0.05),见图2B。


图1进正常大鼠脊髓表面给予IL-1β不影响C纤维介导的基本突触传递,也不影响高频刺激诱导的脊髓背角LTP
3p38MAPK和NF-κB信号通路在IL-1β诱导的脊髓背角LTP中的作用
在给IL-1β之前60 min,脊髓表面给予p38 MAPK抑制剂SB203580或NF-κB 抑制剂PDTC或JNK抑制剂SP600125, 结果显示,SB203580 (100 μmol/L, 200 μL) 和PDTC (10 pmol/L, 200 μL)可以阻断IL-1β诱导的LTP,而SP600125 (100 μmol/L,200 μL)未能阻断IL-1β诱导的LTP,见图3。这表明IL-1β通过激活p38 MAPK和NF-κB信号通路诱导神经损伤大鼠脊髓背角LTP。
讨 论
1致炎症细胞因子在脊髓LTP和海马LTP中作用不同
大量文献表明病理浓度的TNF-α 和IL-1β可以抑制高频刺激诱导的海马LTP[13-14]。而现在我们的研究发现脊

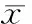
图2神经损伤大鼠脊髓表面给予IL-1β可以诱导C纤维诱发电位的LTP

图3p38MAPK和NF-κB信号通路而不是PDTC通路参与IL-1β诱导的LTP
髓背角给予IL-1β既不影响正常大鼠脊髓的基本突触传递也不影响高频刺激诱导的脊髓LTP,但是在神经损伤(SNI或L5 VRT)引起的病理性疼痛大鼠上可以诱导出脊髓LTP。IL-1β对脊髓背角可塑性的影响和TNF-α 几乎相同。这些结果都支持我们的推测,即致炎细胞因子对海马和脊髓背角突触可塑性的影响完全不同。在海马和脊髓背角都存在LTP现象,海马是学习记忆的结构基础,海马LTP也是学习记忆的突触模型;而脊髓背角是感觉传入的第一级换元部位,脊髓LTP是病理性疼痛的突触模型。以往的研究发现脊髓LTP和海马LTP存在许多相同机制,而我们的研究发现了IL-1β在两种LTP中的作用完全相反,以IL-1β为靶点治疗病理性疼痛不会损害海马LTP,从而可以避免损害学习记忆功能。
2致炎症细胞因子对突触可塑性作用的机制
有文献报道生理浓度的TNF-α引起神经元表面AMPA (alpha-amino-3-hydroxy-5-methl-4-isoxalzole propionic acid)受体表达增加,从而增加突触传递效率[15]。白细胞介素-1受体基因敲除小鼠的海马记忆功能受损,并且海马CA1区和齿状回的LTP完全消失[16]。这些数据表明海马记忆功能和长时程突触可塑性都需要TNF-α和IL-1的参与。然而,病理浓度的TNF-α 和 IL-1β通过激活p38 MAPK、JNK 和 NF-κB信号通路抑制海马LTP[17-19]。在脊髓背角TNF-α通过激活p38 MAPK、JNK 和 NF-κB信号通路诱导神经损伤大鼠的脊髓背角LTP[20]。我们现在的研究发现p38 MAPK 和 NF-κB 的抑制剂可以完全阻断IL-1β诱导的脊髓LTP,但JNK的抑制剂没有此效果。这表明JNK信号通路对TNF-α诱导的LTP很重要,但是并不参与IL-1β诱导的LTP。
3外周神经损伤时致炎症细胞因子的来源
有研究表明,外周神经损伤时,脊髓背角胶质细胞(包括小胶质细胞和星形胶质细胞)大量激活,活化的胶质细胞合成并释放致炎细胞因子(包括TNF-α 、IL-1β、IL-6等),参与神经病理性疼痛的产生和维持[21]。我们早期的研究也发现,脊髓背角小胶质细胞的活化是诱导脊髓LTP的必要条件[22],因此推测脊髓背角的胶质细胞通过致炎细胞因子参与脊髓LTP及病理性疼痛的诱导和维持。
综合已有的研究和本实验结果,我们认为海马LTP和脊髓LTP机制并不完全相同,致炎症细胞因子TNF-α 和IL-1β在2种LTP中的作用完全相反,而外周神经损伤时,活化的胶质细胞合成并释放致炎细胞因子,因此以胶质细胞及致炎细胞因子为靶点的药物可以选择性抑制脊髓LTP,治疗病理性疼痛的同时不损害海马的学习记忆功能。
[1] Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus[J]. Nature,1993, 361(6407): 31-39.
[2] Liu XG, Sandkühler J. Long-term potentiation of C-fiber-evoked potentials in the rat spinal dorsal horn is prevented by spinalN-methyl-D-aspartic acid receptor blockage[J]. Neurosci Lett, 1995, 191(1-2): 43-46.
[3] Zhang HM, Zhou LJ, Hu XD, et al. Acute nerve injury induces long-term potentiation of C-fiber evoked field potentials in spinal dorsal horn of intact rat[J]. Sheng Li Xue Bao, 2004, 56(5): 591-596.
[4] Ji RR, Kohno T, Moore KA, et al. Central sensitization and LTP: do pain and memory share similar mechanisms?[J]. Trends Neurosci, 2003, 26(12): 696-705.
[5] Yang HW, Hu XD, Zhang HM, et al. The roles of CaMKII, PKA and PKC in the induction and maintenance of LTP of C-fiber evoked field potentials in rat spinal dorsal horn[J]. J Neurophysiol, 2004, 91(3): 1122-1133.
[6] Huang YY, Kandel ER. D1/D5 receptor agonists induce a protein synthesis-dependent late potentiation in the CA1 region of the hippocampus[J]. Proc Natl Acad Sci U S A, 1995, 92(7): 2446-2450.
[7] Zhou LJ, Zhong Y, Ren WJ, et al. BDNF induces late-phase LTP of C-fiber evoked field potentials in rat spinal dorsal horn[J]. Exp Neurol, 2008, 212(2): 507-514.
[8] Bellinger FP, Madamba S, Siggins GR. Interleukin 1 β inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus[J]. Brain Res, 1993, 628(1-2): 227-234.
[9] Katsuki H, Nakai S, Hirai Y, et al. Interleukin-1 β inhibits long-term potentiation in the CA3 region of mouse hippocampal slices[J]. Eur J Pharmacol, 1990, 181(3): 323-326.
[10]Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain[J]. Pain, 2000, 87(2): 149-158.
[11]Li L, Xian CJ, Zhong JH, et al. Effect of lumbar 5 ventral root transection on pain behaviors: a novel rat model for neuropathic pain without axotomy of primary sensory neurons[J]. Exp Neurol, 2002, 175(1): 23-34.
[12]Chaplan SR, Bach FW, Pogrel JW, et al. Quantitative assessment of tactile allodynia in the rat paw[J]. J Neurosci Methods, 1994, 53(1): 55-63.
[13]Tancredi V, D’Arcangelo G, Grassi F, et al. Tumor necrosis factor alters synaptic transmission in rat hippocampal slices[J]. Neurosci Lett, 1992, 146(2): 176-178.
[14]Murray CA, Lynch MA. Evidence that increased hippocampal expression of the cytokine interleukin-1β is a common trigger for age- and stress-induced impairments in long-term potentiation[J]. J Neurosci, 1998, 18(8): 2974-2981.
[15]Beattie EC, Stellwagen D, Morishita W, et al. Control of synaptic strength by glial TNFα[J]. Science, 2002, 295(5563): 2282-2285.
[16]Avital A, Goshen I, Kamsler A, et al. Impaired interleukin-1 signaling is associated with deficits in hippocampal memory processes and neural plasticity[J]. Hippocampus, 2003, 13(7): 826-834.
[17]Coogan AN, O’Neill LA, O’Connor JJ. The P38 mitogen-activated protein kinase inhibitor SB203580 antagonizes the inhibitory effects of interleukin-1β on long-term potentiation in the rat dentate gyrusinvitro[J]. Neuroscience, 1999, 93(1): 57-69.
[18]Curran BP, Murray HJ, O’Connor JJ. A role for c-Jun N-terminal kinase in the inhibition of long-term potentiation by interleukin-1β and long-term depression in the rat dentate gyrusinvitro[J]. Neuroscience, 2003, 118(2): 347-357.
[19]Albensi BC, Mattson MP. Evidence for the involvement of TNF and NF-κB in hippocampal synaptic plasticity[J]. Synapse, 2000, 35(2): 151-159.
[20]Liu YL, Zhou LJ, Hu NW, et al. Tumor necrosis factor-alpha induces long-term potentiation of C-fiber evoked field potentials in spinal dorsal horn in rats with nerve injury: the role of NF-kappa B, JNK and p38 MAPK[J]. Neuropharmacology, 2007,52(3): 708-715.
[21]Watkins LR, Maier SF. Beyond neurons: evidence that immune and glial cells contribute to pathological pain states[J]. Physiol Rev, 2002, 82(4): 981-1011.
[22]钟 祎,黄阳亮,许继德.酪氨酸家族激酶在大鼠脊髓背角LTP中的作用及其机制[J]. 中国病理生理杂志, 2011,27(4):727-731.
Roleofinterleukin-1βinspinalLTPinratswithneuropathicpain
ZHONG Yi1, HUANG Yang-liang2, XU Ji-de1
(1DepartmentofPhysiology,GuangzhouMedicalCollege,Guangzhou510182,China;2DepartmentofSpineSurgery,TheFirstAffiliatedHospital,SunYat-senUniversity,Guangzhou510700,China.E-mail:victoria0720@126.com)
AIM: To investigate the role of interleukin-1β (IL-1β) in the long-term potentiation (LTP) of C-fiber-evoked field potentials in rats with neuropathic pain.METHODSThe rat model of neuropathic pain was produced by spared nerve injury (SNI) of sciatic nerve or the method of lumbar 5 ventral root transection (L5 VRT). The effect of exogenous IL-1β on C-fiber-evoked field potentials of spinal dorsal horn was tested in both intact rats and the rats with neuropathic pain. The roles of p38 MAPK and NF-κB in the process were also evaluated.RESULTSIL-1β at concentration of 500 μg/L affected neither basal synaptic transmission mediated by C-fiber nor spinal LTP induced by high frequency stimulation in intact rats. However, low concentration (5 μg/L) of IL-1β induced LTP of C-fiber-evoked field potentials in the rats with neuropathic pain. Pretreatment with either p38 MAPK inhibitor (SB203580) or NF-κB inhibitor (PDTC) completely blocked LTP induced by IL-1β.CONCLUSIONExogeneous IL-1β might induce spinal LTP of C-fiber-evoked field potentials in the rats with neuropathic pain. p38 MAPK and NF-κB may be involved in the process.
Interleukin-1; Long-term potentiation; p38 MAPK; NF-kappa B
R338
A
10.3969/j.issn.1000-4718.2012.03.028
猜你喜欢
中国疼痛医学杂志(2022年9期)2022-10-08
作文周刊·小学二年级版(2022年20期)2022-05-05
感染、炎症、修复(2021年1期)2021-07-28
昆明医科大学学报(2021年2期)2021-03-29
昆明医科大学学报(2021年2期)2021-03-29
妇女(2020年4期)2020-04-20
创新作文(小学版)(2019年10期)2019-09-25
中成药(2018年12期)2018-12-29
中国病理生理杂志(2015年8期)2015-12-21
中国应用生理学杂志(2014年3期)2014-01-22
