
双标记13C,15N3-呋喃妥因的制备
2012-10-24 01:17徐建飞杜晓宁
化学研究 2012年4期
徐建飞,杜晓宁,王 伟,张 彰
(1.上海大学 环境与化学工程学院,上海200444; 2.上海化工研究院 上海稳定同位素工程技术研究中心,上海200062)
双标记13C,15N3-呋喃妥因的制备
徐建飞,2,杜晓宁2,王 伟2,张 彰1*
(1.上海大学 环境与化学工程学院,上海200444; 2.上海化工研究院 上海稳定同位素工程技术研究中心,上海200062)
以双标记13C,15N3-氨基脲为原料,先与苯甲醛缩合,继而与氯乙酸乙酯取代、环化,再经盐酸水解反应后与5-硝基糠醛二乙酯反应,最终制得双标记13C,15N3-呋喃妥因.产物经红外光谱、高效液相色谱及质谱表征.结果表明,所选用的合成路线反应条件温和,产物总收率高于60%,且同位素丰度不下降;目标产物的化学纯度>99.0%,13C同位素丰度>98%,15N同位素丰度>99%.
氨基脲;13C;15N;双标记呋喃妥因;制备
呋喃妥因,化学名为1-{[(5-硝基-2-呋喃基)亚甲基]氨基}-2,4-咪唑烷二酮,最初由美国 Norwich Eaton制药公司研制[1],属硝基呋喃类广谱抗菌药物.早期曾被广泛用作动物饲料添加剂,后发现其具有较强的细胞诱变性和动物致癌毒性[2],因而在饲料生产中遭到全面禁用[3].另外,鉴于呋喃妥因也能作为化妆品添加剂,为避免含有此类药物的化妆品给消费者带来危害,我国《化妆品卫生规范》[4]中明确规定这种药物在化妆品中为禁用物质.为了对饲料和化妆品中的呋喃妥因进行检验,常需使用双标记13C,15N3-呋喃妥因作为内标物,通过同位素稀释质谱法(IDMS)[5-6]进行定量的分析测定.
目前,同位素标记的呋喃妥因合成方法还未见报道,但非标记呋喃妥因的制备方法主要有以下两种:1)JULIAN等[7]采用呋喃与1-氨基海因反应,中间体再经混酸硝化后,即得呋喃妥因;2)章思规[8]和JACK[9]将水合肼与尿素缩合,再在碱性条件下与氯乙酸乙酯缩合等多步反应制得.但采用上述方法合成13C和15N双标记呋喃妥因均存在不足.前一方法引入同位素标记很难,且产生大量的废酸;而后一方法则需使用大量同位素标记的水合肼,成本较高.综合上述两种合成方法,作者以自制的13C,15N3-氨基脲为原料[10],经苯甲醛保护后,先与氯乙酸乙酯缩合环化,所得中间体经盐酸水解后再与5-硝基糠醛二乙酯反应,最终制得13C,15N3-呋喃妥因的双标记化合物.该方法操作简单,副产物少,产物收率高于60%,且没有同位素丰度的稀释现象.
1 实验部分
1.1 药品与仪器
13C,15N3-氨基脲(自制),氯乙酸乙酯(Sigma-Aldrich公司),5-硝基糠醛二乙酯(百灵威化学试剂有限公司),呋喃妥因标样(TCI),其余均为市售的国产试剂.
数显熔点仪 WRS-1B(上海精密科学仪器有限公司),Magna-IR-550型红外光谱仪(Nicolet Co.),WATERS1525型高效液相色谱仪(WATERS Co.),同位素质谱计(Finnigan Co.),LC-MS(Thermo Electron Co.).
1.2 制备方法
13C,15N3-呋喃妥因的合成路线见图1.

图1 13 C,15 N3-呋喃妥因的合成路线(*为同位素标记位置)Fig.1 The synthesis route of 13 C,15 N3-nitrofurantoin(*is the isotope labeled)
1.2.1 苯亚甲基氨基脲(Ⅱ)的制备
在三口瓶中加入苯甲醛(0.1mol)和氨基脲盐酸盐(0.1mol),50%的乙醇水溶液作为溶剂,50℃下反应1h.反应结束后,经过滤、水洗、醇洗和真空干燥,得到白色固体(Ⅱ),收率93%.m p 214~215℃;ESIMSm/z:164.1[M+H]+,186.0[M+Na]+.
1.2.2 苯亚甲基氨基乙内酰脲(Ⅲ)的制备
将0.1mol苯亚甲基氨基脲(Ⅱ)置于500mL三口瓶中,加入乙醇钠(0.1mol)的无水乙醇溶液(50 mL).滴加氯乙酸乙酯30mL,反应回流数小时.将反应物浓缩至干,用稀碱液溶解固体并过滤除去不溶物,滤液用稀盐酸调节pH=6~7,过滤收集固体.真空干燥后得到白色粉末(Ⅲ),收率83%.m p 254~255℃;ESI-MSm/z:204.6[M+H]+,226.1[M+Na]+.
1.2.3 1-氨基乙内酰脲盐酸盐(Ⅳ)的制备
将苯亚甲基氨基乙内酰脲(Ⅲ)15g加入到50mL盐酸溶液(6mol/L)中,加热并回流.反应过程中同时进行水蒸气蒸馏,以除去苯甲醛.反应液经活性炭脱色、蒸干后,用乙醇重结晶,得到白色结晶物(Ⅳ),收率85%.m p 199~200℃;ESI-MSm/z:116.1[M+H]+.
1.2.4 呋喃妥因(Ⅴ)的制备
在装有回流冷凝管、温度计的三口瓶中,加入1-氨基乙内酰脲盐酸盐1.5g,水10mL.再缓慢滴加2.5g 5-硝基糠醛二乙酯的乙醇溶液10mL,在冰水浴下缓慢加入浓硫酸3mL,加热反应2h.结束后冷至5~10℃,逐渐有黄色固体析出,过滤后固体用水洗至中性,再用乙醇洗涤.用DMF重结晶得到黄色固体(Ⅴ),50℃下真空干燥4h,收率92%.m p 258~261℃(分解);IR:(KBr),σ/cm-1:3 624.7,3 479.2(NH),3 141.3,3 019.0(C-H),1 732.4(═C O),1 614.7(═C C),1 437.3(NO2).ESI-MSm/z:237.15[M-H]-.
1.2.5 高丰度标记物合成实验
以自制高丰度13C,15N3-氨基脲为原料,替代天然丰度氨基脲,按上述方法合成13C,15N3-呋喃妥因,收率60%,13C丰度98.10%,15N丰度为99.08%,化学纯度为99.0%.ESI-MSm/z:241.14[M-H]-.
1.3 分析方法
纯度分析采用液相色谱方法[11-12]分析13C,15N3-呋喃妥因.HILIC C-18柱,流动相:A(CH3OH)/B(H2O)/C(HCOOH)=26∶73∶1,柱温30℃,检测波长265nm,流速1mL/min.采用MAT271质谱计,用微量法制备样品[13-14],分析其13C,15N的同位素丰度.LC-MS分析离子源为ESI,模式为正或负离子模式.
2 结果与讨论
2.1 苯亚甲基氨基脲(Ⅱ)的合成
2.1.1 溶剂、温度的影响
实验选用了不同的溶剂和反应温度,以考察它们对中间体—苯亚甲基氨基脲(Ⅱ)收率的影响.实验操作方法参见1.2.1,实验结果见表1.
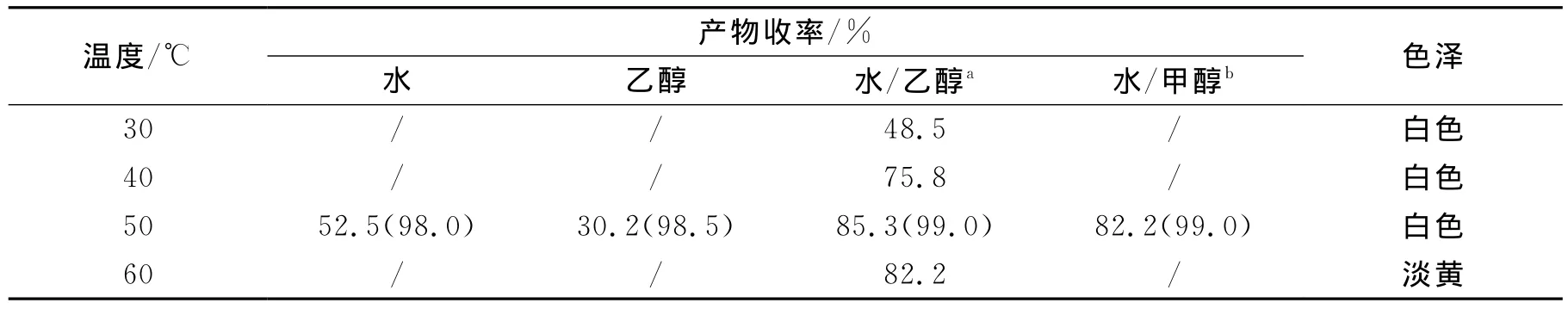
表1 温度、溶剂对中间体(II)产率的影响Table 1 Effect of temperature and solvent on yield of intermediate(II)
由表1可以看出,用纯水、乙醇作溶剂时,因苯甲醛在水中溶解度较差,氨基脲盐酸盐在乙醇中的溶解度非常低,致使收率偏低.采用水/醇(V/V=1∶1)混合溶剂时,则收率有明显的提高,且以水/乙醇(V/V=1∶1)为溶剂时最佳.从反应温度看,随着反应温度的升高,收率也逐步升高,但反应温度达60℃时,产物收率趋于恒定,且产品颜色偏黄,可能是副产物所致,故选择反应温度以50℃更为适宜.
2.1.2 反应时间的影响
另外,我们还就反应时间对中间体(Ⅱ)收率的影响进行了探索.在50℃下,以水/乙醇1∶1(V/V)为混合溶剂,分别选取反应时间为0.5、1.0、2.0、3.0和4.0h进行了实验比较,其结果见表2.
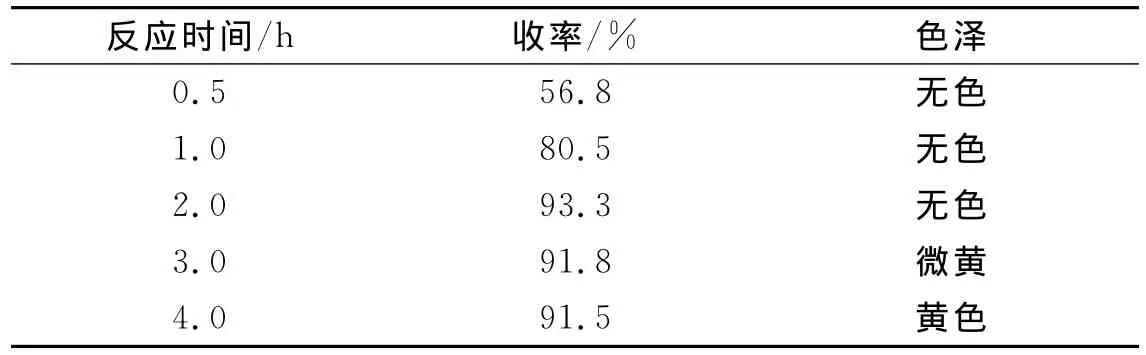
表2 反应时间对中间体(Ⅱ)收率的影响Table 2 Effect of reaction duration on yield of intermediate(II)
从表2可以看出,收率随着反应时间的延长而增加,但超过3h后,产物外观变黄,这可能是副反应所致,故选择反应时间2h为宜.
2.2 1-氨基乙内酰脲盐酸盐(Ⅳ)的合成
根据上述合成路线,第3步为酸性水解反应,其目的是脱除保护基,而制得中间产物—1-氨基乙内酰脲盐酸盐(Ⅳ).实验过程中,就体系酸浓度、反应时间对产物收率的影响作了相应研究,实验操作步骤参见1.2.3,实验结果见表3.
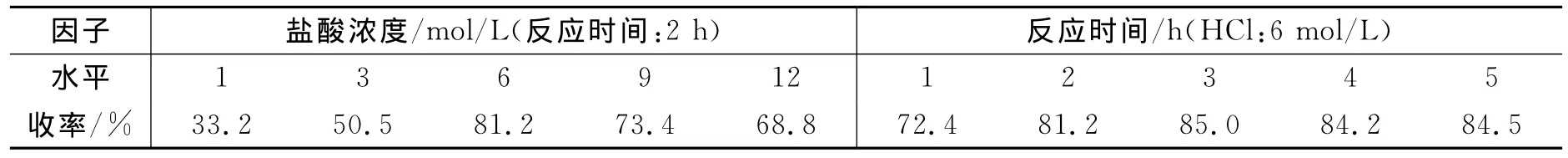
表3 酸度、反应时间对1-氨基乙内酰脲盐酸盐(Ⅳ)收率的影响Table 3 Effect of acid concentration,reaction duration on yield of AHD.HCl(Ⅳ)
显然,酸的浓度过低时,不能将1-氨基乙内酰脲盐酸盐全部水解;浓度过高时,则容易发生串联副反应,而导致产物的收率下降,所以选择6mol/L最为适宜.另一方面,在反应初期,产物收率随时间延长而增大;当达到3h后,产物收率基本趋于恒定,约85%.
2.3 正交实验
呋喃妥因(Ⅴ)合成反应所涉及的影响因素较多,包括5-硝基糠醛二乙酯和1-氨基乙内酰脲的摩尔比(A)、反应温度(B)、反应时间(C)等,为此本实验采用L9(34)正交表进行实验探索,并以呋喃妥因产物收率作为考察指标,对其反应工艺条件作优化处理,实验结果见表4.

表4 L9(34)的正交实验表及极差RTable 4 Dates and range Rof L9(34)
从表4中R值的直观分析可以看出,各因素对呋喃妥因收率的影响程度是:原料摩尔比(A)>反应温度(B)> 反应时间(C).原料摩尔比(A)对呋喃妥因的收率有着极为显著的影响,而反应时间对呋喃妥因的收率则几乎没有影响,由正交表中结果可知,合成呋喃妥因反应的最优化条件是:原料摩尔比为1.1∶1.0,反应温度为60℃,反应时间取3h.
另外,根据正交实验结果进行实验验证,其呋喃妥因收率为92.0%,纯度99.0%以上,完全符合正交实验结果.
2.4 产物的结构表征
2.4.1 HPLC
以市售天然丰度的呋喃妥因试剂为外标,通过HPLC的测试分析,证明本实验所合成的产物为13C,15N3-呋喃妥因,其纯度为99.0%.
2.4.2 LC-MS
实验就合成产物—13C,15N3-呋喃妥因进行了质谱测试分析.根据结构其标记分子量为242,负离子模式为[M-H]-即241.谱图显示m/z=241.14与理论值完全相符,因此,可确定合成所得的标记样品为13C,15N3-呋喃妥因.
2.4.3 MAT 271同位素质谱的丰度测定
实验使用微量法制备样品,用MAT271质谱计测得:13C,15N3-呋喃妥因的13C同位素丰度为98.10%,15N同位素丰度为99.08%.原料氨基脲的15N同位素丰度为99.26%,13C同位素丰度为98.40%,其同位素稀释效应的影响小于0.5%.
根据以上HPLC、MS和MAT271同位素质谱分析的测试结果,可以确定本实验合成所得化合物的结构与13C,15N3-呋喃妥因完全一致,且同位素丰度未被稀释.
结论:设计并合成了双标记13C,15N3-呋喃妥因化合物,并对反应工艺作了详细的探索.通过单因素实验和正交设计实验得出合成最优化工艺条件,中间体苯甲醛缩氨基脲的收率达93.0%,经氯乙酸乙酯缩合成环反应、盐酸水解反应,再与5-硝基糠醛二乙酯反应,最终制得目标产物—13C,15N3-呋喃妥因,总收率60%以上.另一方面,合成产物经HPLC、MS与同位素质谱分析等测试,测试结果显示实验制得的产物与设计的目标产物分子结构完全一致,且化学纯度>99.0%,13C同位素丰度>98.10%,15N同位素丰度>99.08%,同位素稀释效应影响小于0.5%.
双标记13C,15N3-呋喃妥因的合成制备,对饲料和化妆品中添加剂的检测提供了分析依据,对呋喃类内标试剂的国产化研究及我国食品和化妆品安全体系的建立,均具有着十分重要的意义.
[1]戚雪勇,吴修艮.呋喃妥因的合成新工艺[J].化学世界,2007,48(4):252.
[2]祝伟霞,刘亚风.动物性食品中硝基呋喃类药物残留检测研究进展[J].动物医学进展,2010(2):99-102.
[3]张 庆,王 超,王 星,等.高效液相色谱法同时测定化妆品中的呋喃妥因和呋喃唑酮[J].色谱,2009(2):237-239.
[4]中华人民共和国卫生部.化妆品卫生规范 [S].北京:中华人民共和国卫生部,2007,28:42.
[5]刘卫霞,罗 勇,杨维成.有机同位素稀释质谱法在食品安全分析中的应用[J].化学世界,2011(3):184-187.
[6]周 霄,王 军,张丽娟.同位素稀释质谱法测量环境样品中的微量元素[D].北京:中国科学院上海冶金研究所,2000.
[7]JULIANG M,NORWICH N Y.Nitrating an N-(2-furyl)alkylidene hydrazing compound:US,2898335[P].1959-08-04.
[8]章思规.精细有机化学品技术手册[M].北京:科学出版社,1991:20-21.
[9]JACK D H,SUTNO G H.Preparation of 1-aminohydrantoin derivatives:US,2990402[P].1961-06-27.
[10]王 伟,杜晓宁,刘占峰,等.13C,15N-盐酸氨基脲合成方法的优化[J].同位素,2010(4):206-210.
[11]MOTTIER P.Quantitative determination of four nitrofuran metabolites in meat by isotope dilution liquid chromatography-electrospray ionisation-tandem mass spectrometry[J].J Chromatogr A,2005,1067(1/2):85-91.
[12]祝伟霞,杨冀州,魏 巍,等.高效液相色谱-串联质谱法测定动物性食品中硝基呋喃类代谢物残留[J].现代畜牧兽医,2008(1):47-50.
[13]杜晓宁,宋明鸣,赵 诚,等.13C-尿素的同位素丰度检测方法[J].同位素,2010(1):40.
[14]杜晓宁,宋明鸣,赵 诚.质谱检测用15N样品的处理方法[J].原子能科学技术,2009(12)增刊:60.
Preparation of double labeled13C,15N3-nitrofurantoin
A novel method was established to synthesize double labeled13C,15N3-nitrofurantoin.Starting material13C,15N3-semicarbazide was firstly converted to benzaldehyde semicarbazone(II)by condensation with benzaldehyde.Compound(II)was then allowed to react with ethyl α-chloroacetate in the presence of sodium ethoxide,forming 1-benzylideneaminohydratoin(III).Compound(III)was hydrolyzed with hydrochloric acid and condensed with 5-nitro furfural diacetate,generating the target compound,nitrofurantoin-(13C,15N3)(V).As-synthesized target product was characterized by infrared spectrometry,high-performance liquid chromatography,and mass spectrometry.It has been found that the established synthetic route is dominated by mild reaction conditions and gives rise to the final product in a yield of above 60%,while the abundance of the isotopes does not tend to decline.Namely,the chemical purity of the target product is above 99.0%,and the abundances of13C and15N are above 98%and 99%,respectively.
nitrofurantoin;13C;15N;double labeled semicarbazide;preparation
O 621.3
A
1008-1011(2012)04-0001-05
2012-02-21.
徐建飞(1979-),男,硕士生,研究方向为化学工程与工艺.*
.E-mail:zhzhang9@shu.edu.cn.
猜你喜欢
能源化工(2021年2期)2021-12-30
科学与财富(2021年33期)2021-05-10
汽车零部件(2018年5期)2018-06-13
广东石油化工学院学报(2016年6期)2016-05-17
电源技术(2015年7期)2015-08-22
合成化学(2015年5期)2015-03-26
同位素(2014年3期)2014-06-13
同位素(2014年2期)2014-04-16
同位素(2014年2期)2014-04-16
石油化工应用(2014年2期)2014-03-11
