
关注血管干细胞与糖尿病并发症——血管干细胞在糖尿病并发症发病与治疗中扮演的角色
2012-10-22 02:28:58基茨
糖尿病天地(临床) 2012年11期
基茨 等
(邓超 编译中南大学糖尿病中心/中南大学湘雅二医院内分泌科)
慢性糖尿病并发症
糖尿病是一种慢性退行性的代谢性疾病,目前还没有治愈手段。当前,糖尿病患者总数接近2.21亿。仅仅在北美,超过10%的人群患有糖尿病。这产生令人惊讶的经济负担,预计2020年加拿大的花费将达到170亿美元/年,美国高达1,160亿美元/年。尽管北美的发病率令人十分担忧,但由于较差的并发症管理和较低的健康管理标准,近80%的糖尿病相关死亡发生在中低等收入国家。尽管已做出极大努力处理这一疾病,WHO提出糖尿病相关死亡在2030年时将会超过现在的两倍。
糖尿病领域最为重要的发现则是1921年胰岛素的问世。外源性胰岛素显著减少了糖尿病性昏迷和酮症酸中毒,拯救了上百万人的生命。然而,糖尿病患者仍然不能免于慢性并发症的伤害。这些长期并发症表现为微血管(视网膜病变、神经病变、肾脏病变和心肌病变)和大血管(动脉粥样硬化)病变。尽管并发症的临床特征各不相同,根本的原因在于目标器官的血管功能失常。两项大型的临床试验为更好理解糖尿病并发症病因做出铺垫,即分别在1993年和1997年完成的糖尿病控制和并发症试验(DCCT)和英国前瞻性糖尿病研究(UKPDS)。在两个试验中,1型和2型糖尿病患者都进行强化血糖控制;这两种情况中,都延迟和(或)抑制了糖尿病并发症的发生。其他因素,如高血脂和高胰岛素血症,也可能参与糖尿病并发症的发病机制。这两项临床试验的结果和多年的糖尿病动物模型及培养细胞研究均证实了这一概念,即高血糖是长期糖尿病后微血管和大血管病变的主要原因。
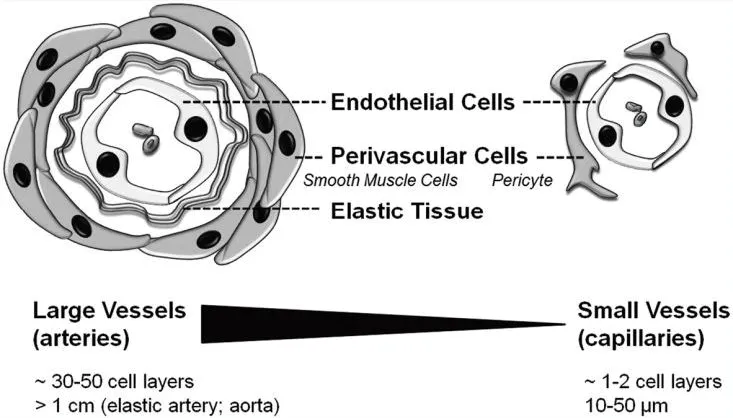
图1 大血管与小血管图解
糖尿病并发症血管功能障碍的分子基础
血管内皮细胞是血管的关键组成部分。这些专门的细胞形成血管的内衬,位于基膜上面,被支持性的血管周围细胞(周细胞或平滑肌细胞)所环绕(见图1)。血管内皮细胞不仅作为一道屏障,在循环血液和灌注组织间产生一道分界面,而且在组织功能及器官发生中起到突出的作用。这些细胞参与多种重要的血管功能,如调节血流量、血压、渗透性、血液流速、血栓形成/纤溶平衡以及白细胞运输。
基于在血管中的解剖学定位,血管内皮细胞首先接触循环中葡萄糖。葡萄糖转运蛋白(Gluts)促进葡萄糖的吸收。血管内皮细胞中主要的Glut是Glut1。尽管Gluts以典型地组织特异性方式表达,在正常生长条件下Glut1无处不在地表达。不像许多其他葡萄糖转运蛋白,Glut1活性和表达水平不随增加或减少的血糖水平而改变。这表明高血糖可能对血管内皮细胞具有极大的有害作用,因为葡萄糖吸收可能不会被活性调节。然而,某些确定的条件下可能会改变Glut1表达。例如,低氧增加内皮细胞Glut1水平。这可能是糖尿病并发症无法控制的内皮细胞功能障碍机制中的其中一项。
体外研究表明,暴露于高水平的葡萄糖导致成熟血管内皮细胞生化改变。这些变化表现为增加的细胞外基质蛋白,如胶原和纤维蛋白和增加的促凝蛋白血管性血友病因子(vWF)及改变的细胞活性。除了增殖和迁移减少之外,许多研究证明高血糖可以直接地促进内皮细胞凋亡。大多认为这一凋亡途径是由氧化应激增加、细胞内钙离子内流增加、线粒体功能障碍、细胞内脂肪酸代谢改变、有丝分裂原激活蛋白激酶(MAPK)信号途径激活和蛋白激酶B(Akt)磷酸化/激活受损所引起的。发生在任一靶器官血管系统结构改变之前最早期的功能改变之一,是内皮依赖性的血管舒张功能受损。这一功能受损因为两种内部调节的机制:血管舒张因子生成减少和血管收缩因子生成增加。在高糖条件下培养的血管内皮细胞中发现,一氧化氮减少和最强力的内皮血管收缩因子——内皮素-1增加。我们和其他人已经证明参与NO生成的酶在接受高糖处理的内皮细胞中上调。然而,酶联反应的解偶联和可能由于氧化应激引起的NO隔离导致NO显著减少。另一得到确认的导致糖尿病中内皮细胞损害增加的途径是氧化应激。高血糖的内皮细胞产生活性氧(ROS)如羟基、过氧化物阴离子和过氧化氢。ROS过量产生可能归因于交替的代谢/信号途径激活,如多元醇途径和氨基己醣途径,以及通过蛋白激酶C、AGE形成和PARP激活(见图2)。这些途径之间均可以彼此加强,以增加ET-1活性,减少NO生物利用度、氧化应激和内皮细胞功能障碍。引人注目地是,我们见到高糖处理的内皮细胞的生化改变与糖尿病患者出现的慢性并发症存在关联。
我们知道改变的内皮细胞为糖尿病患者长期血管功能障碍提供了支柱(图 3)。内皮细胞结构和功能的改变导致随后的血管网络失常,组织则将出现血流较少和缺血的迹象。正常而言,在这些情况下会出现适应性反应。尽管依赖于参与的器官而症状不同,糖尿病患者都会出现血管适应性反应。例如,视网膜和肾脏典型地表现为血管形成增加,然而这一过程在心脏和下肢中受损。糖尿病对器官靶点选择性损伤提示组织微环境和内皮细胞本质特点两者都具有重要性。
生长因子和细胞外基质(ECM)蛋白是维持新血管形成和疤痕形成/纤维化之间平衡的两种主要的调节因子。血管内皮生长因子(VEGF)是一种内皮细胞特异性促有丝分裂原,在许多疾病模型中促进血管生成。糖尿病患者心脏血管生成缺乏的同时,也发现心肌细胞VEGF和它的受体表达减少。这与视网膜VEGF水平升高形成直接对比,伴随难以控制的视网膜新生血管生成。除了生长因子外,ECM调节血管细胞,可能导致糖尿病并发症中高血糖水平有差别的效应。内皮细胞表面整合素结合到ECM蛋白调节细胞存活/凋亡、生长和细胞支架的改变。因此,血管形成高度依赖于血管单位的细胞组分和环绕的支架蛋白之间的交互作用。事实上,被认为在肿瘤发生中促进新血管生成ECM改变在视网膜血管发展中是类似的,包括纤连蛋白和层粘连蛋白增加。糖尿病动物的视网膜基膜早至糖尿病发病8周后出现一种类似的蛋白成分——升高的胶原蛋白IV、纤连蛋白和层粘连蛋白。伴随ECM蛋白异质性,心脏中ECM沉积增加可能导致血管生成反应受损。心脏大量出现的成纤维细胞,可能通过功能性的ET-1导致ECM蛋白沉积。体外实验表明ET-1通过成纤维细胞增加ECM组分生成。
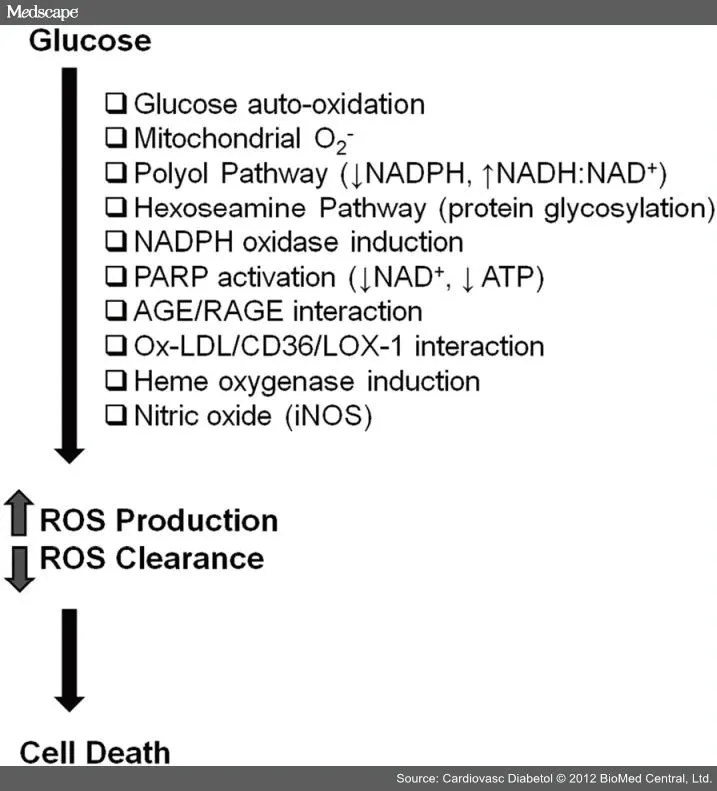
图2 ECs中葡萄糖诱导的氧化应激机制
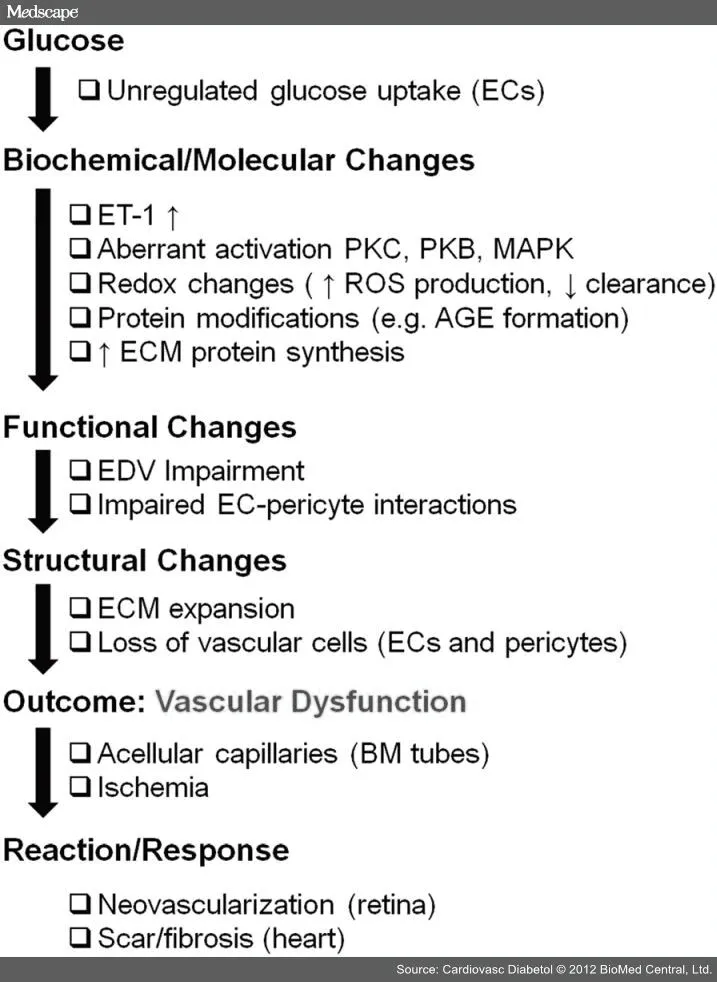
图 3 糖尿病个体中导致血管损伤的机制
不考虑器官系统,血管内皮细胞是高血糖损害的主要媒介,他们发生功能和结构上的改变。随后出现的血管调节受损、渗透性增加、ECM扩增和整个血管网络功能障碍引起流动到器官的血液减少,进而导致难以控制的并发症进展。为了阻断这些并发症的进展和修复损伤,我们将需要替换损伤的内皮细胞或创造崭新的血管网络。考虑到逐渐增加的包括血管壁在内的多种组织干细胞或祖细胞证据,我们可以推测糖尿病患者出现修复机制受损的原因是这些干/祖细胞也同样受到影响。
血管干细胞(VSCS):目前的证据和希望
干细胞被定义为通过自身具有自我更新和分化为功能性成熟细胞的能力。这些细胞的潜力由他们的层级和特殊化水平决定。干细胞已在多种胚性组织中发现,包括骨髓、血液、脂肪和皮肤。发现这些干细胞群为非侵入性组织修复和包括血管组织在内的组织再生提供机会。为了再生成全新的血管网络(重新形成),我们必须首先发现合适的细胞来源。理想情况下是一种细胞类型或亚群可以生成内皮细胞和支持性的管周细胞。一种能生成成熟/功能性的血管细胞的特异性血管干细胞(VSC)的概念逐渐得到发展。几个研究组已经在小鼠和人体研究中证明一种常见的血管前体细胞的存在。Kattman等在小鼠中使用细胞追踪研究,显示心肌细胞来源于一种表达VEGF受体-2(VEGFR2/Flk1)的细胞群,提示这群细胞来源于一种具有血管分化潜力的祖细胞。他们接着在胚胎干细胞分化模型中进行这些研究,他们从这一模型中分离人类拟胚体心血管的祖细胞(brachyury+; VEGFR2+),成功地证明其有产生心肌细胞、内皮细胞和血管平滑肌细胞的潜力。Yamashita等的研究表明,来源于胚胎干细胞的VEGFR2+细胞,通过不同的培养环境可以分化为内皮细胞和壁细胞。当置于真实的培养系统中,这些细胞也能够生成血管组织。同样地,Ferreira等的研究表明,来源于EBs血管祖细胞(CD34+)会产生内皮和平滑肌样细胞,当植入体内时有能力形成血管网络。
VSCs准确的特性目前尚不清楚。有充足的证据说明这些VSCs存在于骨髓和循环中,并且不同于造血干细胞。从循环中选择CD133+细胞纯化一群细胞,在不同培养条件下会生成家系限制性的内皮祖细胞(EPCs)和间充质/中胚层祖细胞(MPCs)。然而,表达造血标志CD45+的细胞不能产生内皮细胞。目前尚不清楚是一种还是多种干细胞亚型属于CD133+阳性这一可以生成内皮细胞和间叶细胞的亚群。更为重要的是干细胞来源的EPCs和MPCs形成功能性血管网络。最近人们提出这是否是一个治疗糖尿病可行的方法。
内皮祖细胞和糖尿病并发症
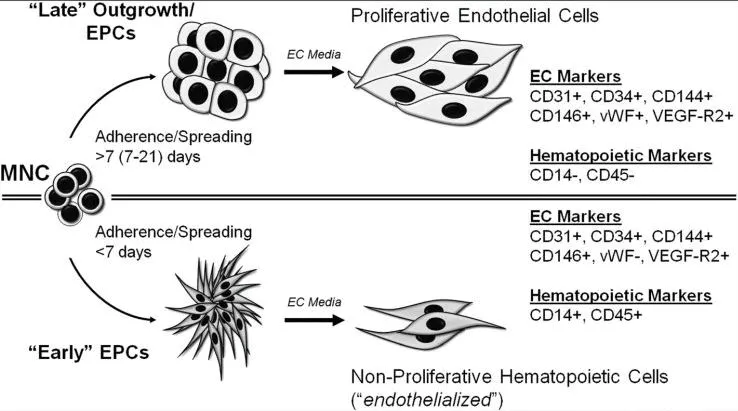
图4 早期与晚期EPCs
祖细胞或前体细胞是家系限制性的,干细胞高度增殖的衍生物。这些细胞可以每10~15小时自我复制增殖翻倍。例如,EPCs具有干细胞和成熟内皮细胞家系的两种标志。由于EPCs具有的潜在用途,临床上或作为生物标志物,精确的识别和可再生的分类极具重要性。尽管在该研究上有所进步,尚没有一致意见应该如何定义EPCs。传统上,EPCs被认为是从外周血或骨髓来源的单个核细胞分离后培养2~4天出现的纺锤状或多形细胞(见图 4)。这些细胞以荆豆凝集素结合和Dil标记的乙酰化低密度脂蛋白吸收为特征。这两组特点被认为是ECs的功能特征。然而,伴随表达一些EC标志物,这些短期的EPC克隆也同样表达单核细胞特异性标志CD14和(或)造血干细胞标志CD45。而且,乙酰化低密度脂蛋白吸收是1979年就已鉴定出来的单核细胞的特征。荆豆凝集素是一种植物凝集素,通过驻留的海藻糖结合到ECs表面。而且,这些驻留的海藻糖不是特异性结合到ECs。
那么什么是EPCs?EPCs最为重要的特征或功能属性是整合到血管的能力。换句话说,EPCs是形成血管的。相比之下,早期的EPCs可能被认为是形成血管的,因为它们通过生长因子加工促进血管形成。我们以及其他人已经广泛描述了血管源性的EPCs标志物表达和细胞活动。EPCs具有ECs和单能性祖细胞的特征。EPCs在CD133(在EPCs阳性表达但培养后丢失)表达上,增殖或生长动力学(EPCs群体倍增时间较短和更高的群体倍增数),以及对内皮抑制素反应(EPCs受到刺激而成熟ECs受到抑制)不同于成熟的ECs。久而久之,EPCs就标志物表达和细胞活性方面上类似于成熟ECs。在血管生成和血管源性的EPCs的许多争论通过进行功能上的细胞活性检测(总结在图5)得到否定。这些评估包括内皮特异性标志物表达,细胞因子处理活性,以及最为重要的是细胞生成血管的能力。
EPCs可能参与慢性糖尿病并发症的血管功能障碍。与健康对照相比,已知1型和2型糖尿病患者含有较低水平的循环EPCs。在两项相似的研究中,流式分析用于定量检测糖尿病患者EPCs(CD34+/VEGFR2+/CD31+)。这些研究显示EPCs分别减少44%和40%。最近发现2型糖尿病患者循环血液中CD34+/VEGFR2+细胞数目与血糖控制相关。这项研究同时指出糖尿病患者循环CD34+/VEGFR2+细胞和动脉硬化之间存在负相关。由于这些表面标志物不是EPCs特有的,减少的数目可能包括造血干/祖细胞水平改变。事实上,一项包括120例缺血性心脏病的大型研究显示骨髓来源的CD34+/CD45+ 细胞减少,这也与糖化血红蛋白水平相关联。体外使用早期EPCs的实验研究同样显示糖尿病患者中EPCs较低者的血管生成能力和粘附到内皮细胞单层的能力受损。关于晚期血管源性EPCs的研究工作极少,我们已知高水平血糖不会改变细胞生长、增殖或晚期EPCs迁移。然而,同样的情况,成熟ECs的ET受体表达增加和血糖诱导的成熟EC凋亡增强。这些发现表明血管源性的EPCs可能抵抗高血糖带来的副作用。
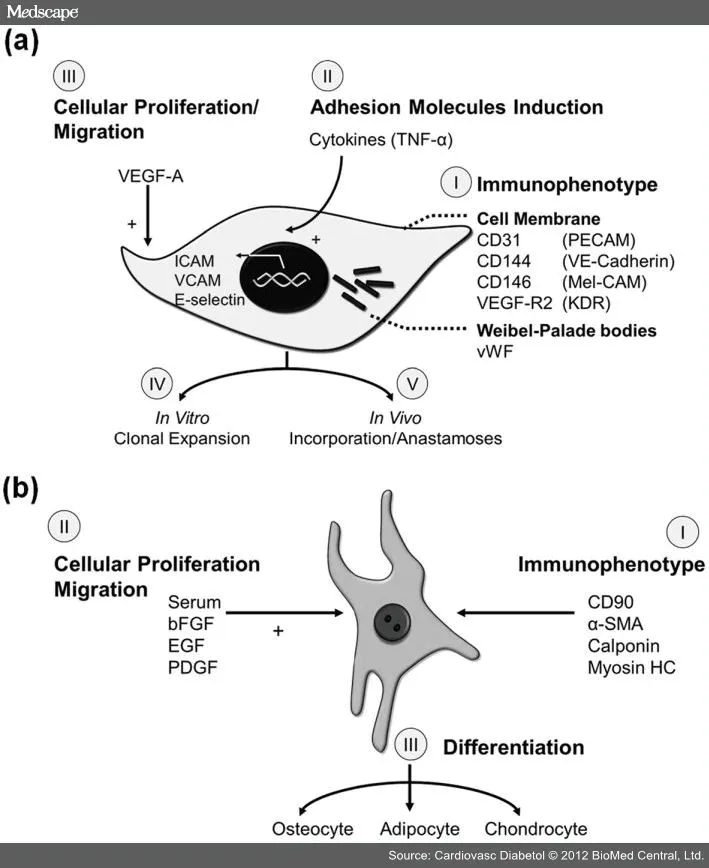
图5 EPCs(a)与MPCs(b)的特征图示
尽管在长病程的糖尿病患者中EPC数目可能会减少,他们仍然存在治疗的希望。如果EPCs的细胞活性在糖尿病中保持完整,体外给予扩增的EPCs应该基本上改善血管功能障碍。不仅是成人血来源的EPCs在体外成功扩增,它们形成完整功能的血管网络能力也在体外得到验证。我们注意到为了形成稳定和持续的血管网,EPCs需要共同移植许多管周细胞。如同EPCs,来源于同样的CD133+部分的MPCs,可能是这一治疗的合适人选。
间充质祖细胞(MPCS)和糖尿病并发症
MPCs是多能细胞,和EPCs一样来源于循环细胞中CD133+亚群。MPCs可以从成人骨髓中大量分离。此外,已经在肝脏、脾脏和脂肪组织中发现MPCs。如同EPCs和其他祖细胞类型,MPCs具有干细胞和成熟细胞的双重特征。MPCs兼具表型和功能特征,包括细胞表面标志物表达、细胞粘附分子和分化潜能(见图 5)。因为没有一种标志物特异性表达于MPCs,适当地鉴别这群细胞亚群时所有的参数都必须考虑到。培养中间充质细胞典型表型为纺锤样形态,然而,依赖于组织来源不同,特别是来源于不同种属时也存在一些异质性。MPCs不表达内皮细胞标志物CD31和造血的标志物CD45。mRNA和(或)蛋白分析可以用于说明平滑肌肌动蛋白、钙调蛋白、CD90、PDGFR和NG2表达。功能上,MPCs分化为间叶系细胞,包括脂肪细胞、骨细胞和软骨细胞。
关于MPCs在糖尿病并发症中可能的致病性作用所知甚少。然而,就已知的治疗性益处,最近的研究显示并发症得到改善,包括心肌病、肾病、神经病变和创伤治疗。使用糖尿病心肌病大鼠模型,静脉给予MPCs,通过增加基质金属蛋白酶(MMP)-2和减少MMP-9改善心肌重塑以及提高心肌功能。此外,我们发现VEGF、胰岛素样生长因子(IGF)-1、肾上腺髓质素和肝细胞生长因子减少。MPCs分化为心肌细胞和血管内皮细胞,改善糖尿病患者心肌灌注和再生。MPCs也成功地改善小鼠糖尿病肾病进展。全身性注射后,前体细胞转移到受损的肾脏,分化为肾脏细胞,改善肾功能以及肾小球结构的再生。进一步,当肌肉内注射时,MPCs通过增加血管源性细胞因子如bFGF和VEGF改善糖尿病多神经病变。在皮肤创伤修复模型中,对链脲霉素诱导的糖尿病大鼠给予MPCs使得延迟的创伤修复时间回归正常。该效应部分是由CD45+细胞浸润到伤口减少所介导的。这项研究涉及正常的MPCs(如细胞分离于非糖尿病大鼠),问题在于糖尿病是否引起MPCs功能上的改变。这是一个所知不多的新的研究领域。然而,近期一项研究表明AGEs(图 2)可能增加活性氧的生成,减少MPCs的增殖和迁移。这是否在人类糖尿病或糖尿病并发症动物模型中发挥作用,仍需要进一步的研究。
考虑到MPCs具有超过其他细胞类型的优势(分化潜能和免疫调节能力),它们可能是糖尿病并发症很好的治疗候选方案。近来在动物模型的研究确实显示了希望。几项研究报告MPCs治疗可以通过旁分泌作用加强血管形成。旁分泌作用可能涉及血管源性因子的释放来促进EPC归巢以及血管网的重建。
结论
检查糖尿病的长期影响已经引起血管内皮细胞作为高血糖诱导损伤的主要靶点的启示。随后,靶器官的整个血管网出现功能障碍,为我们在糖尿病患者所见到的并发症提供了基础。实验证据表明,分离于糖尿病小鼠的干/祖细胞能够重建血管稳态。这提示糖尿病主要的干细胞缺陷是数量减少。这种减少可能发生在骨髓和循环之间的某处。如果我们能找到一种方法利用这两种细胞类型来修复血管损伤和重建血管功能,就有希望改善慢性并发症(见图 6)。
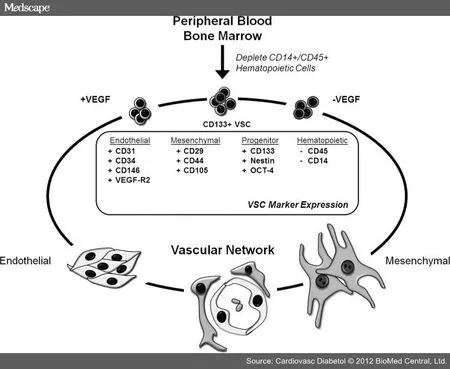
图6 VSCs治疗糖尿病的潜力
治疗性血管形成的成功依赖于多种因素,其中之一是工程血管与宿主血管形成稳定的和功能性的吻合能力。目前使用人类脐静脉内皮细胞以及人类微血管内皮细胞已成功获得了新生血管。然而,由于较低的收益,临床上使用这些特别的内皮细胞类型存在限制。考虑到EPCs比较容易从成人外周血分离出来,可以非侵入性方法获得这些细胞并在体外大量扩增。扩增后,这些细胞被移植到糖尿病患者体内重建血管稳态。先前已经说明成人和脐带血来源的EPCs有能力在体内形成功能性血管网。尤其重要的是,为了维持稳定的功能性网络,这需要移植管周细胞。MPCs是合适的候选细胞,因为它正如EPCs,是可靠来源的管周细胞。它们能够从骨髓甚至成人外周血等位点以最小的并发症风险被分离出来。我们先前已经报道皮下共同植入EPCs和MPCs到无胸腺小鼠的背部,产生了与宿主循环系统形成功能性吻合的人类微血管。
猜你喜欢
今日农业(2022年13期)2022-09-15 01:21:20
生物学通报(2020年10期)2020-08-13 08:52:26
中国眼镜科技杂志(2019年9期)2019-11-11 12:15:32
知识经济·中国直销(2017年10期)2017-11-07 02:39:52
安徽医科大学学报(2016年12期)2017-01-15 14:21:48
中国病理生理杂志(2015年8期)2015-12-21 12:38:16
安徽医科大学学报(2015年9期)2015-12-16 11:09:42
医学研究杂志(2015年11期)2015-06-10 06:44:03
中国卫生(2014年2期)2014-11-12 13:00:14
云南中医学院学报(2014年5期)2014-07-31 18:00:10
