
单体、寡聚体及纤维状Aβ毒性作用的比较研究
2012-10-09 03:56:14孔繁军崔德华
中国实验诊断学 2012年9期
孔繁军,陈 强,周 亮,崔德华*
(1.北京海淀医院 神经内科,北京100080;2.宁波市北仑宗瑞医院 神经内科;3.北京大学医学部 神经科学研究所)
阿尔茨海默病(Alzheimer disease,AD)是一种发生于老人的最常见的神经退行性疾病,其主要病理学特征则是在脑中形成大量的老年斑和神经原纤维缠结以及出现弥漫性脑萎缩[1]。其病因发病机制的研究1984年,Glenner and Wong从老年斑中纯化和分离得到Aβ并解读得到其蛋白序列[1],才开始有了显著的进展。随后的研究表明老年斑的主要成分是β-淀粉样蛋白(β-amyloid peptide,Aβ),由β-淀粉样蛋白前体(amyloid precursor protein,APP)裂解产生,Aβ的寡聚体和纤维化是AD患者脑内重要的病理改变,在凝聚形成Aβ纤维的过程中,具有过氧化损伤,引起炎症反应,损伤突触功能等多种神经毒性,对AD的病理进程起关键作用[2]。本实验采用单体、寡聚体及纤维化Aβ损伤大鼠海马CA1区来制备AD大鼠模型,并采用流式细胞术检测AD大鼠不同脑区细胞凋亡情况。
1 材料与方法
1.1 实验动物 选取雄性Wistar大鼠64只,鼠龄3-4个月,体重200-250g,大鼠随机分为Aβ组、生理盐水组及假手术对照组,每组16只。
1.2 不同种类 Aβ制备[5]将 Aβ1-40溶于灭菌过的超纯水,配成500μmol·L-1储存液分装,于-20℃冻存。寡聚体Aβ使用前于37℃孵育48h,纤维状Aβ使用前于37℃孵育21d。不同类型Aβ使用AFM法观察确认寡聚体Aβ和纤维状Aβ。单体Aβ1-40不在37℃孵育7d,冻结后马上使用。采用核黄素标记光度法测定Aβ凝聚度。
1.3 动物模型的制作 用10%水合氯醛腹腔注射(0.3-0.35ml/100g)麻醉大鼠。将头部固定在立体定位仪上,头背中部纵向切口,暴露颅骨,参照《大鼠脑立体定位图谱》,选右侧海马为注射区,定位坐标:前囟后3.0mm,中线右侧2.0mm,硬膜下2.6 mm。钻开颅骨,分离暴露硬脑膜,垂直进针至靶点,缓慢注射5μl寡聚体 Aβ1-40(浓度为1μg/μl,预先37℃下孵育1w),注射时间为5min,留针5 min,保证溶液充分弥散,撤针,缝合切口,所有操作均在无菌条件下进行。假手术组切开硬脑膜后即缝合伤口。Aβ组、生理盐水组及假手术对照组用生理盐水按等量代替。
1.4 不同Aβ对海马细胞凋亡 (1)制备单细胞悬液:术后7d各组取5只大鼠,处死动物,在冰盘上快速取新鲜海马、额叶、颞叶约0.2g,0.1MPBS(pH7.4)洗涤,剪碎,0.3%胰蛋白酶3.0ml,37℃消化30min,在PBS中洗涤、吹打成单细胞悬液,200目尼龙网过滤,-20℃预冷的无水乙醇3ml中固定,4℃保存备用。(2)细胞凋亡百分率测定[5]:细胞数调整至1×106个,加100μl RNAse 37℃水浴30min,加入PI染色液800μl混匀,4℃避光30 min,上机进行FCM检测,测定数据按FAC Scan所配置的ModFit软件进行分析,以细胞周期各时相以及AP区亚峰细胞百分率记录数据。
1.5 采用核黄素标记光度法测定Aβ凝聚方法 参照我们已发表JBC论文的方法测定Aβ凝聚[5]。
2 结果
2.1 不同的Aβ的β凝聚度
Aβ随时间依赖性凝聚度增强。单体Aβ与寡聚体Aβ组间数值间有显著差异(P※※<0.01),并且纤维化Aβ与寡聚体Aβ组间数值间有显著差异(P※※<0.01),见图1。
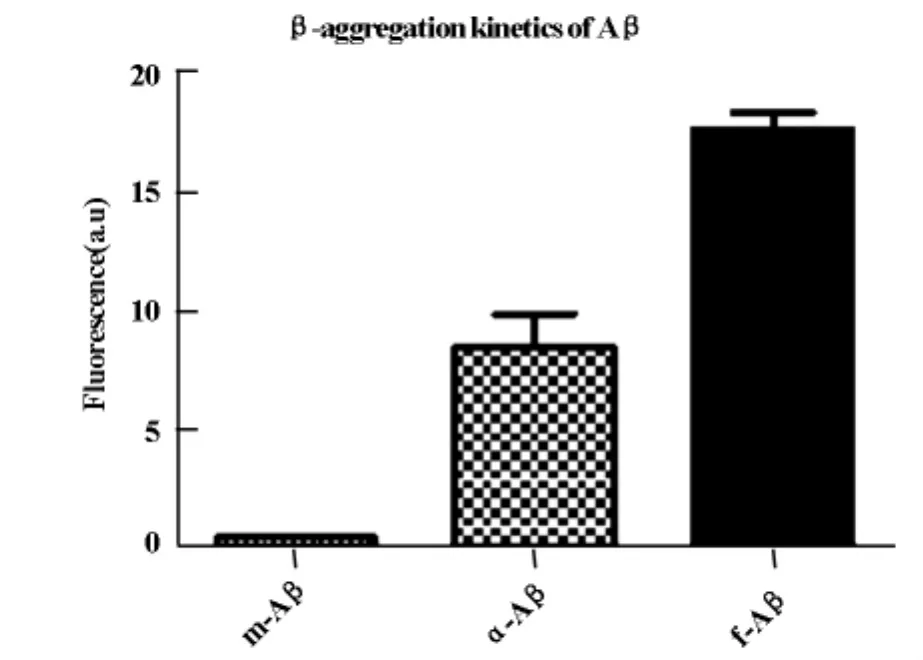
图1 不同的Aβ的β凝聚度
2.2 不同的Aβ对海马细胞凋亡的变化 对照组与单体Aβ组间无统计学差异(P>0.05);多聚体Aβ组与单体及纤维化Aβ、对照组数值间有显著差异(P※※<0.01),寡聚体Aβ组海马含G1期细胞百分数明显升高,见图2。
此结果说明单体Aβ没有毒性,寡聚体Aβ对海马神经细胞毒性最大,但是转化成纤维化Aβ,则神经毒性反而降低。

图2 表示不同的Aβ对海马细胞的毒性作用。mo-Aβ,单体Aβ;o-Aβ,寡聚体Aβ;f-Aβ,纤维化 Aβ,各组n=5,**P<0.01。
2.3 不同的Aβ的β凝聚度和海马神经细胞的死亡之间的关系
为了阐明Aβ的β凝聚性对神经细胞的毒性之间的关系,对单体Aβ,寡聚体Aβ及纤维化Aβ组的凝聚度和细胞毒性做了相关分析。结果显示Aβ凝聚度与海马神经元细胞毒性无正相关,即并不是凝聚度越高毒性越大,反而纤维化Aβ毒性下降。此结果进一步支持寡聚体Aβ在AD发生起关键作用。
3 讨论
AD是一种导致老年性痴呆的常见疾病,其临床特征主要为进行性记忆减退、言语和行为障碍,AD的病因和发病机制还不十分清楚。但目前认为Aβ是AD老年斑的核心成分,被认为是多种原因导致AD的共同通路[1]。我们采用经体外孵育的凝聚状态的Aβ1-40海马CA1区立体定向注射建立AD动物模型,以往的实验结果表明单侧海马CA1区注射后1周的Aβ组大鼠明显表现出记忆获得和记忆再现的损伤作用,形态学观察证明在Aβ脑内可引起神经元的凋亡,说明Aβ海马区注射Aβ1-40建立的模型比较符合AD的自然病理过程。
本实验采用FCM以碘化丙啶(PI)为特异性荧光探针标记细胞,单参数分析细胞内DNA相对含量,对Aβ1-40海马CA1区损伤大鼠不同脑区脑组织所制作的单细胞悬液进行凋亡百分率检测,结果提示,Aβ组凋亡百分率明显升高,与其它各组比较差异明显,说明Aβ神经毒性可致神经细胞凋亡性改变。
脱氧核苷酸转移酶以模板依赖性方式催化用荧光素或同位素标记的脱氧核苷酸与DNA片段游离3-OH末端发生聚合反应,用于标记DNA链断片,然后进行或同位素检测。TUNEL法实际上是分子生物学与形态学相结合的研究方法,对完整的单个凋亡细胞核或凋亡小体进行原位染色,能准确地反应细胞凋亡最典型的生物化学和形态学特征,可检测出极少量的凋亡细胞,灵敏度较高[2]。
Aβ与细胞凋亡关系密切[3],在培养的神经细胞中加入Aβ后神经元出现了细胞凋亡典型形态学和生物化学特征[6]。在APP717转基因小鼠脑内Aβ广泛沉积,大量神经元发生凋亡,同时伴有P53蛋白表达的增加[7]。将 Aβ1-40注射到成年小鼠脑内,海马可见到明显细胞丢失,而在Caspase-3基因缺失小鼠,则细胞丢失不明显[8]。本实验结果用FCM和TUNEL法证明在Aβ脑内可引起神经元的凋亡,支持细胞凋亡参与了AD发病的观点。
AD发病机制中涉及细胞凋亡有关的多个环节[9]。老年斑的主要成分寡聚体Aβ激活Caspases,一组启动和执行凋亡的蛋白水解酶,导致细胞核和细胞骨架蛋白的裂解,其中Tau蛋白的降解是神经纤维变性的关键[10]。Aβ的神经毒性作用可直接或间接损伤线粒体膜而使膜电位下降,PT孔的开启可以引发一系列与细胞凋亡相关的重要事件[11],如线粒体内Ca2+的释放及质子的渗漏、细胞色素C的释放、Caspases激活因子的释放、细胞内氧化还原状态的改变,Bcl-2家族促进和抑制凋亡蛋白的参与等。不同信号的传导最终集中到线粒体上来启动或抑制这些事件及其效应的产生[12]。
Aβ寡聚体比单体及纤维化Aβ毒性显著增高,因此,防止Aβ寡聚体化可抑制凋亡蛋白的产生,减轻细胞凋亡的发生,可能对减轻神经系统的损害提供新的思路和策略。
[1]Cotman CW,Poon WW,Rissman RA,et al.The role of caspase cleavage of tau in Alzheimer disease neuropathology[J].J Neuropathol Exp Neurol,2005,64(2):104.
[2]Wu CK,Thal L,Pizzo D,et al.Apoptotic signals within the basal forebrain cholinergic neurons in Alzheimer's disease[J].Ecp Neurol,2005,195(2):484.
[3]Yanker BA.Mechanisms of neuronal degeneration in Alzheimer’s disease[J].Neuron,1996,16:921.
[4]Chui DH,Tanahashi H,Ozawa K,et al.Transgenic mice with Alzheimer presenilin 1mutations show accelerated neurodegeneration without amyloid plaque formation[J].Nature Medicine,1999,5(5):560.
[5]Yoshiike Y,Chui DH,Akagi T,et al.Specific compositions of amyloid-beta peptides as the determinant of toxic beta-aggregation[J].J Biol Chem,2003,278(26):23648.
[6]Alvarez AR,Godov JA,Mullendorff K,et al.Wnt-3aovercomes beta-amyloid toxicity in rat hippocampal neurons[J].Exp Cell Res,2004,297(1):186.
[7]Laferla FM,Hall CK,Ngo L,et al.Extra cellular deposition ofβamyloid upon P53-dependent neuronal cell death in transgenic mice[J].J Clin Invest,1996,98(7):1626.
[8]Takuma H,Tomiyama T,Kuida K,et al.Amyloid beta peptideinduced cerebral neuronal loss is mediated by caspase-3in vivo[J].J Neuropathol Exp Neurol,2004,63(3):255.
[9]Clementi ME,Pezzotti M,Orsini F,et al.Alzheimer's amyloid beta-peptide(1-42)induces cell death in human neuroblastoma via bax/bcl-2ratio increase:An intriguing role for methionine 35[J].Biochem Biophys Res Commun,2006,342(1):206.
[10]Wirths O,Multhaup G,Bayer TA.A modified beta-amyloid hypothesis:intraneuronal accumulation of the beta-amyloid peptide--the first step of a fatal cascade[J].J Neurochem,2004,91(3):513.
[11]Keil U,Bonert A,Marques CA,et al.Amyloid beta-induced changes in nitric oxide production and mitochondrial activity lead to apoptosis[J].J Biol Chem,2004,279(48):50310.
[12]Green DR,Reed JC.Mitochondria and apoptosis[J].Science,1998,281:1309.
猜你喜欢
保健与生活(2019年17期)2019-11-08 02:13:40
养生阅刊(2019年7期)2019-09-10 07:22:44
海峡姐妹(2019年6期)2019-06-26 00:52:48
黄河黄土黄种人(2018年5期)2018-06-07 08:30:54
天然产物研究与开发(2018年3期)2018-05-07 06:38:37
中成药(2017年9期)2017-12-19 13:34:27
CHINESE JOURNAL OF AERONAUTICS(2017年1期)2017-11-21 12:54:14
中成药(2017年5期)2017-06-13 13:01:12
河南大学学报(医学版)(2014年1期)2014-03-30 12:09:53
首都医科大学学报(2013年6期)2013-10-25 09:36:54
