
价层电子对互斥理论中两个问题的讨论
2012-09-25 07:02张晨曦王雪峰
大学化学 2012年6期
张晨曦 王雪峰
(同济大学化学系 上海 200092)
价层电子对互斥(VSEPR)理论认为分子构型取决于价层电子对的构型,并且由于Pauli不相容原理价层电子对在空间应该尽量远离[1-3]。由于不依附于复杂的量子化学计算以及轨道模型,仅通过简单的Lewis电子式对分子构型进行判断,VSEPR理论在教学中获得了广泛应用。
然而,在VSEPR理论的教学过程中,笔者发现许多教材对于多重键方面的论述存在误区[4-15],即许多教材使用σ-π成键理论解释VSEPR理论在含有多重键分子构型方面的结论,而这些结论却是基于Pauli的弯键理论建立的。因此,本文着重通过对C2H4分子的讨论,阐述VSEPR理论讨论含有多重键分子构型的理论基础;另外,介绍了含有5个电子对的AL5分子的特殊性质,并提出运用该构型分子判断电子对相互排斥作用的大小。
1 VSEPR理论有关多重键的讨论
在现今教材中讨论中心原子与配体之间通过多重键(即双键和三键)结合时,均指出共价双键和共价三键都当作一个共价单键来处理,每个键只计算其中的一对σ电子,但是教材中并没有给出多重键的价层电子对被作为单键处理的解释。另外,在现今教材和教学过程中,几乎都忽视了VSEPR理论解释多重键的基础——Pauli的弯键理论,而是均采用了σ-π成键理论进行解释[4-14]。虽然VSEPR理论仅通过价层电子对之间Pauli斥力判断分子构型,不涉及共价键的形成过程和键的稳定性,但是在多重键的讨论中,VSEPR理论认为多重键的电子对均居于两个中心原子之间,这也就是Pauli所提出的弯键理论的基础;而σ-π成键理论则认为两个中心原子的p轨道肩并肩重叠形成π键,不仅无法给出多重键看作单键的原因,同时无法解释因多重键而产生的键角偏差。以下分别用σ-π成键理论和弯键理论讨论乙烯分子,简单介绍两种理论并解释因多重键产生键角偏差以及多重键在VSEPR理论中看作单键的原因。
σ-π成键理论认为乙烯分子中的两个中心C原子均采取sp2杂化,分别与2个H原子和1个C原子形成3个σ键,轨道间夹角呈120°;未参与杂化的pz轨道垂直于杂化轨道平面“肩并肩”形成π键。弯键理论则认为中心C原子依旧采取sp3杂化,两个C原子之间有两个价层电子对,形成两个C—C弯键(图1)。
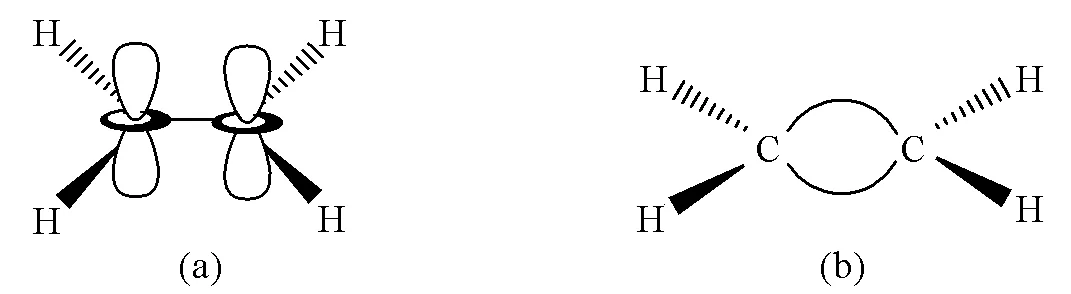
图1 C2H4分子的σ-π模型(a)和C2H4分子的弯键模型(b)
虽然σ-π理论对离域共轭作用以及多重键的光谱给出合理的解释,但是该理论无法解释多重键对键角的影响。通过量子化学计算和实验可得,乙烯分子H—C—H键角小于120°为116.2°[15]。若按照σ-π成键理论,由于pz轨道垂直于杂化轨道平面,“肩并肩”成键的π电子对3个σ键的作用应该是等效的,因此教材中不能说由于π电子影响了H—C—H键角,也不能通过σ-π成键理论推出多重键之间的相互排斥作用大于单键之间的相互排斥作用的结论。又比如F2CCH2分子,其F—C—F键角为110°,更偏离sp2杂化的120°而是接近sp3杂化的109.5°[16]。另外,由于σ-π成键理论认为中心两个C原子中间只形成了一个σ键,而π键是由中心原子p轨道之间重叠形成的,因此σ-π成键理论也无法说明将多重键看作单键的原因。相比之下,Pauli的弯键理论则可以很好地解释多重键键角变化的问题。仍以乙烯为例,VSEPR理论认为中心C原子4个价层电子对由于Pauli斥力呈正四面体构型,其中两个价层电子对同时受到两个C原子核的吸引,使得两个价键相互靠近收拢,而对另外一侧的C—H键的排斥作用减小,使得H—C—H夹角由109.5°增大到116.2°。
本文此处希望说明VSEPR理论是基于弯键模型,弯键模型认为形成多重键是为满足最大程度成键使得两中心原子间形成价键弯曲,键之间是等性的;正因形成了这样的价层电子对,方可以运用VSEPR理论进行解释。这也进一步说明了VSEPR理论和经典价键理论(VB法)有本质上的区别。前者不依附于轨道模型,而是考虑价层电子对的Pauli斥力并通过几何推导得出分子构型。因此,中心原子的杂化轨道形状不影响VSEPR理论的预测,反而在教学中用VSEPR理论先预测分子构型再反推中心原子的杂化类型。Pauli的弯键理论认为由于双键中两个价层电子对以及三键中3个价层电子对被两个中心原子核束缚在一起,多重键成键时必须克服Pauli斥力而且无法满足最大重叠的原则,这也很好地解释了弯键键能比σ键能低的原因。
虽然Pauli的弯键模型能够很好地解释非离域π键以及桥键等相关问题,但是弯键理论也存在很多问题,比如对于离域共轭效应以及多重键的光谱无法给出满意的解释。因此,现今对于大部分多重键的讨论使用σ-π成键理论解释,仅仅对于含有非离域π键以及桥键的小分子多使用弯键模型解释。
故使用Pauli的弯键理论解释VSEPR理论过程中将多重键看作单键的问题时,必须明确VSEPR理论其实运用了电子密度区域的概念将其简化[3]。电子密度区域运用一个球体代表价层电子对,该球形区域表示在该区域价层电子对出现的概率大而在区域之外价层电子对几乎不出现。由于Pauli不相容原理,两个球体不会相互重叠,而且区域越大其相互排斥作用越大。通常在实际运用VSEPR理论预测分子构型时,将双键看作四电子密度区域而将三键看作六电子密度区域。由于双键或三键两核之间有2个或3个电子对,其可看作是一个电子密度区域(一个更大的球体)和其他电子密度区域相互作用,这也是部分教材中将双键和三键看作是单键的原因。但因双键和三键的密度区域大于单键的密度区域,因此电子密度区域相互之间的排斥作用更大。
分析乙烯分子构型可知,由于双键电子密度区域较单键更大,其排斥作用也更大,使得H—C—H键角小于VSEPR推测的120°。而VSEPR理论则认为由于双核对价层电子对的束缚,使得其对C—H价层电子对的排斥作用变小,键角由正四面体的109.5°增大为116.2°。可见,虽然电子密度区域概念和VSEPR理论均是基于Pauli不相容原理,并且电子密度区域概念可以使VSEPR理论更为简单,但是它们在分析多重键时有着很大的区别。而如今很多教材仅仅引用电子密度区域的结论简化了VSEPR理论的使用,却没有进行说明和阐述,这也使得部分学生在多重键的讨论过程中产生了疑问。
笔者建议在VSEPR理论教学过程中加入上述讨论,首先引入电子密度区域理论,讲明其可以将VSEPR理论的应用简单化、形象化;在多重键领域中对比Pauli的弯键理论和σ-π理论,开拓学生视野并引发学生对多重键的进一步思考;最后在电子密度区域和弯键理论的基础上解释VSEPR理论将多重键看作单键的原因。
2 五配体化合物(AL5)的构型
VSEPR理论指出,最稳定的分子构型使得成键和未成键电子对相互排斥作用最小,即在空间价层电子对的几何距离最大。对于AL5分子,通过Pauli斥力的量子化学推导以及空间几何推导可以得出其最稳定的构型是三角双锥构型[9]。具体来说,轴线上的电子对与中心原子所在锥平面上的3个电子对之间的键角呈90°,而锥平面上的电子对仅与轴线上的两个电子对键角呈90°,与锥平面内其他两个电子对的键角均大于120°。因此,在轴线上的电子对与其余电子对相互之间的作用力较锥平面上电子对大。为使整个分子更稳定,AL5分子中的5个A—L键长并不完全相等,轴线上的键长较锥平面内略大[3]。
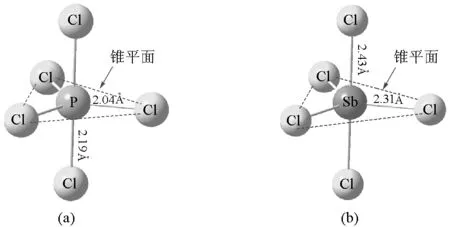
图2 PCl5分子(a)与SbCl5分子(b)
基于价层电子对相互排斥最小的原则,排斥作用大的配体应优先占据锥平面的位置,而排斥作用小的配体占据轴线上的位置。这就为我们提供了一个定性判断电子对相互排斥作用大小的方法。下面使用AL5分子模型分别对未成键孤对电子对,配体电负性以及多重键3个方面进行验证。
2.1 对未成键孤对电子对的讨论
由于成键电子对受到两个原子核的吸引,而未成键的孤对电子对只受一个核的吸引,其电子密度区域更大,因此,孤对电子对之间的相互排斥作用大于成键电子对之间。
在讨论含有孤对电子对的AL5分子构型时可知:为满足价层电子对相互作用最小的原则,价层电子对应该优先占据锥平面上的位置以减小电子对之间的排斥作用。如SF4分子中心S原子的一对孤对电子优先居于锥平面,并且由于其排斥作用较大使得轴线呈微小弯曲;XeF2分子的3对孤对电子也是优先居于锥平面(图3)。

图3 SF4分子构型(a)和XeF2分子构型(b)
2.2 对配体电负性的讨论
教材中很少提及配体电负性对分子构型的影响,但当分子由不同配体构成或含有多重键时,配体的电负性对键角也产生了影响。VSEPR理论指出配体的电负性越大,配体之间的排斥作用越小[3],其中,可以认为孤对电子对的电负性为0。VSEPR理论认为Pauli斥力是价层电子对相互排斥的主要原因,而Pauli斥力随作用距离的增大而迅速减小。因此配体电负性越大,价层电子对离核越远,价层电子对之间的Pauli斥力就越小。在讨论配体电负性对AL5分子构型的影响时可知:电负性越小的配体优先居于锥平面的位置以减小电子对之间的相互排斥作用。以PF4Cl分子为例,Cl原子的电负性小于F原子,因此Cl原子优先取代锥平面上的F形成PF4Cl(图4)。
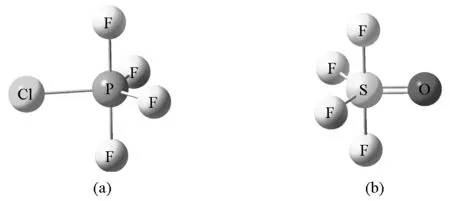
图4 PF4Cl分子构型(a)和SOF4分子构型(b)
2.3 多重键的相互排斥作用

综上所述,AL5以其独特的非完全对称构型为我们提供了定性判断电子对相互作用大小的方法,并分别对含有孤对电子对、不同电负性配体以及多重键分子中的电子对相互作用大小进行比较。笔者建议将这有趣的判断方法加入到相关教材中,让学生对VSEPR理论以及分子构型有更深层次的认识。
3 结语
大学无机化学以及结构化学课程中的价键理论是本科生后续学习化学的基础,而VSEPR理论又因其简单有效而越来越受学生的青睐。本文解释了VSEPR理论在讨论多重键时存在的误区,并对比了Pauli的弯键理论和σ-π成键理论,强调电子密度区域概念在VSEPR理论中的应用;其次,总结了运用AL5分子判断价层电子对之间排斥作用大小的方法。希望借此让学生对VSEPR理论有更深入的理解。
[1] Gillespie R J,Nyholm R S.QRevChemSoc,1957,339
[2] Gillespie R J.Molecular Geometry.London:Van Nostrand Reinhold,1972
[3] Gillespie R J,Hartgittai I.The VSEPR Model of Molecular Geometry.Boston:Allyn and Bacon,1991
[4] 宋天佑.简明无机化学.北京:高等教育出版社,2007
[5] 大连理工大学无机化学教研室.无机化学.第5版.北京:高等教育出版社,2006
[6] 冯传启,杨水金,刘浩文,等.无机化学.北京:科学出版社,2010
[7] 张欣荣,阎芳.基础化学.北京:高等教育出版社,2007
[8] 曹凤岐.大学化学基础.北京:高等教育出版社,2005
[9] 古国榜,李朴.无机化学.北京:化学工业出版社,2007
[10] 北京大学《大学基础化学》编写组.大学基础化学.北京:高等教育出版社,2006
[11] 邵学俊,董平安,魏益海.无机化学.第2版.武汉:武汉大学出版社,1994
[12] 天津大学无机化学教研室.无机化学.第4版.北京:高等教育出版社,2010
[13] 颜秀茹.无机化学与化学分析.天津:天津大学出版社,2004
[14] 宋天佑,程鹏,王杏乔,等.无机化学(上).北京:高等教育出版社,2004
[15] Hirota E,Endo Y,Saito S,etal.JMolSpectrosc,1981,89:223
[16] Mijlhoff F C,Renes G H,Kohatd K,etal.JMolStruct,1977,39:241
猜你喜欢
辽宁科技大学学报(2022年5期)2023-01-04
山西大学学报(自然科学版)(2022年5期)2022-11-23
高中数理化(2022年16期)2022-09-14
国防科技大学学报(2020年6期)2020-12-07
原子与分子物理学报(2020年5期)2020-03-17
测绘通报(2019年11期)2019-12-03
赤峰学院学报·自然科学版(2019年5期)2019-09-10
青岛大学学报(工程技术版)(2019年2期)2019-09-10
中学生数理化(高中版.高考理化)(2019年6期)2019-06-22
考试周刊(2018年39期)2018-04-19
