
长效多肽药物化学修饰的研究进展
2012-09-18 02:19龚梦嘉
重庆理工大学学报(自然科学) 2012年4期
姜 和,龚梦嘉
(1.重庆理工大学 药学与生物工程学院,重庆 400054;2.重庆前沿生物技术公司,重庆 400041)
多肽类药物作为近年来研究的热点,受到国内外制药界的广泛关注,在癌症、艾滋病[1]等重大疾病的治疗中显示出巨大的应用价值。
多肽类药物虽然安全、生物活性好,但多数因半衰期较短而需频繁给药,这使病人的依从性大大降低,并且治疗费用也较大,因此,为了保持药效的持久、不用频繁给药,对于长效多肽类药物的研发近年来成为了多肽药物领域的研究热点。多肽类药物的长效化技术主要有体外化学修饰技术、生物技术和缓控释制剂技术[2]。体外化学修饰技术是在多肽水平上,使用合适的修饰方法和修饰剂对其进行化学修饰[3],使修饰后的多肽类药物在体内的半衰期延长。该技术主要包括主链末端的修饰[4]、侧链的修饰[5]、环化[6]、氨基酸替换[7]、糖基化修饰[8]及 PEG 修饰[9]等。
1 主链末端的修饰
多肽类药物常用的主链末端修饰方法是N端的乙酰化作用和C端的酰胺化作用[10],分别对肽链两端氨基和羧基进行保护,使多肽不会很快地被相应的多肽蛋白酶降解。目前这一技术已经广泛应用于多肽的化学合成中。N端乙酰化通常是在固相合成多肽反应的整个肽链组合完毕后,加入乙酸酐使其乙酰化。C端的酰胺化则是通过选择裂解产物为酰胺的树脂或者选择不同裂解方式来完成。Brinckerhoff等[11]通过对具有免疫原性的多肽MART-I27-35进行N端的乙酰化或者C端的酰胺化,明显提高了该多肽在血浆中的稳定性。
主链末端连接不同长度的脂肪酸、主链C端或N端的PEG修饰及糖基化修饰,其基本原理都是增加多肽分子的相对分子量和空间位阻,提高其对多肽水解酶的稳定性,减少肾小球的滤过作用。RC-160是一种具有抗增生活性的生长激素抑制剂类似物。Dasgupta等[12]将各种长度的脂肪酸连接在RC-160的主链末端。与未修饰的RC-160相比较,修饰后的RC-160对于胰岛素有更高的抵抗能力和更长的半衰期。
2 侧链的修饰
多肽侧链基团的化学修饰是通过选择性试剂(也称修饰剂)与多肽侧链上特定的功能基团发生化学反应而实现,从而改变多肽的化学性质,如消除免疫原性和免疫反应性、延长了多肽药物在体内的作用时间等[13-14]。David Sabatino 等[15]在对生长激素释放多肽(growth hormone releasing peptide,GHRP-6)类似物的合成研究中,通过对Trp2、Ala3、Trp4的化学修饰,在侧链上连接不同的基团(如图1所示),从而形成氮杂多肽(aza-peptide)。在考察经修饰的各种氮杂多肽的IC50时,发现其中一种结构为His-D-Trp-Ala-azaPhe-D-Phe-Lys-NH2的氮杂多肽与受体的结合率是未经修饰的GHRP-6的1000倍。
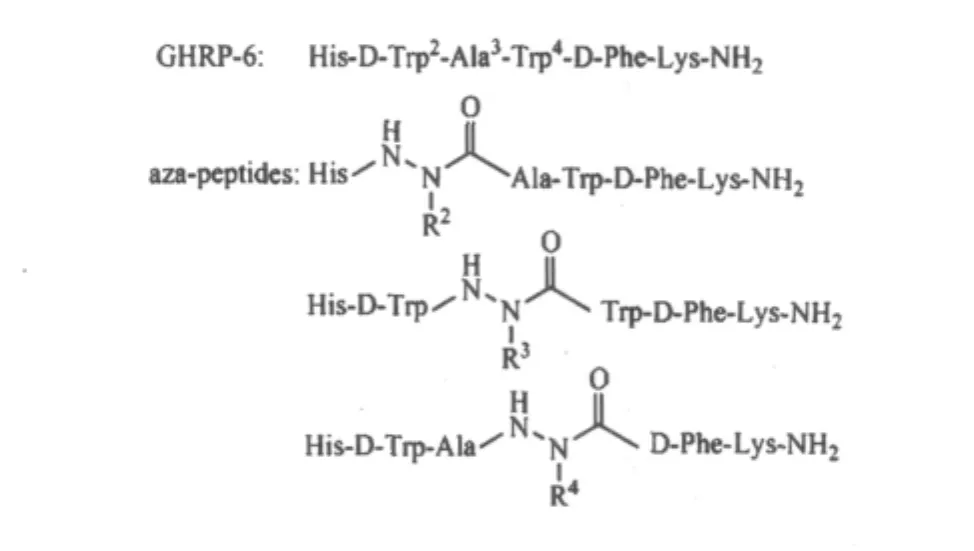
图1 GHRP-6及其类似物结构式
正交保护氨基酸在固相多肽合成中的应用对多肽的侧链修饰起着重要的作用。正交保护是指在一定条件下,分子中的一组保护基可以被选择性地脱除,而其他保护基不受影响,进而达到可选择性修饰的目的[16]。谢东等[17]在合成的长效HIV融合抑制剂艾博卫泰中,对第13位的赖氨酸(Lys)采用 Alloc保护的Fmoc-Lys(Alloc)-OH,用特定的条件选择性地脱除保护基Alloc,而另一氨基保护基Fmoc和肽链上其他氨基酸的侧链保护基均不受影响,再用Fmoc-AEEA(Fmoc-8-氨基-3,6-二氧杂辛酸)和MIPA(3-马来酰亚胺基丙酸)对Lys13进行修饰,使肽链进入血液后能够迅速与白蛋白结合,从而发挥长效的作用。经临床实验验证,艾博卫泰的半衰期可长达2周。
对肽链中某一个氨基酸进行定点修饰是侧链修饰的一项前沿的、重要的技术,也是实现侧链修饰可控的保证。Peng Wu等[18]报道了重组蛋白在哺乳动物细胞中表达,通过基因编码乙醛标记可对重组蛋白中的某些特定位点进行特异性的乙醛标记,这些乙醛标记既可作为定点修饰的位点,也可用于进一步的化学修饰。这项技术已应用在对单克隆抗体、多肽类药物等的定点修饰中。
3 环化
在多数情况下,机体内的氨肽酶及羧肽酶很容易从常见的直链肽的两端进行逐步的切割分解,使直链肽被降解,因此,对肽链进行环化结构改造,可以提高多肽类药物在机体内的生物稳定性,延长半衰期[19]。按照桥头环化位置可将环肽分为5类:C端与N端相连、侧链间相连、N端与侧链相连、C端与侧链相连以及主链上的N-N相连[20],如图2 所示。

图2 按照桥头环化位置的环肽分类
生长调节因子(growth regulating factor,GRF)是由29个氨基酸组成的多肽,肽序为YADAIFTNSYRKVLGQLSARKLL QDIMSR。该多肽在37℃的猪血浆中的半衰期只有15 min。研究人员选取不同位点进行环化处理,得到不同的衍生物,实验结果如表1所示,其半衰期明显提高到4 h以上[21]。
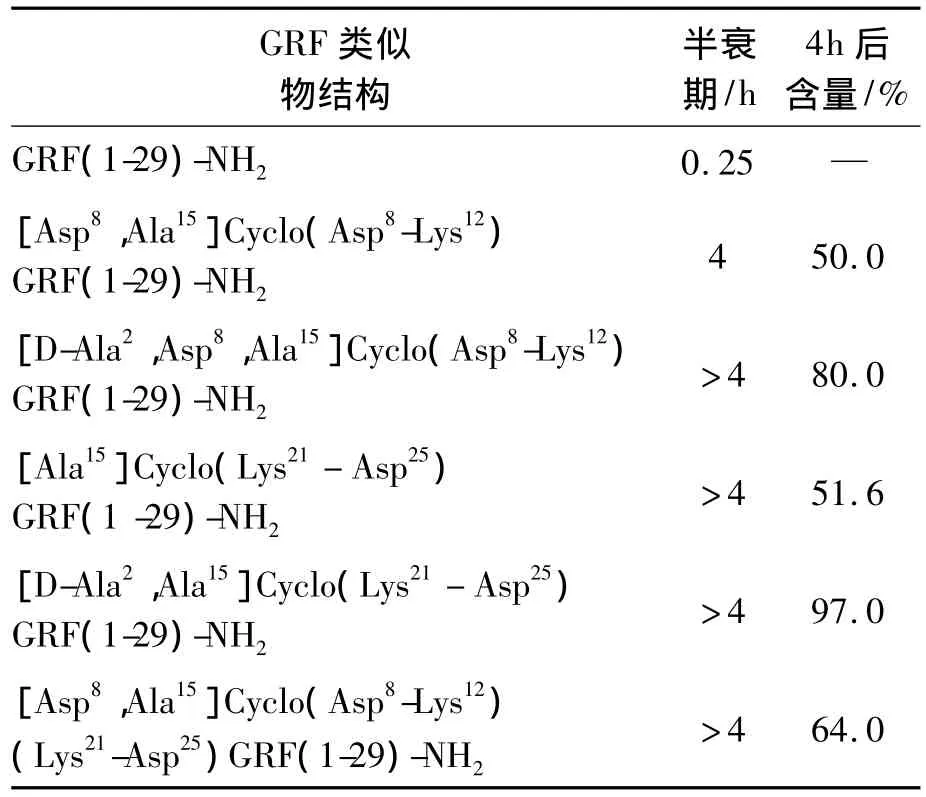
表1 环化修饰后GRF类似物在猪血浆中的稳定性
Annett Rozek等[22]报道了一吲哚类多肽类似物 CP-11,结构为 ILKKWPWWPWR RK-NH2,通过二硫键进行成环修饰,得到cycloCP-11,在胰岛素共同存在和作用下,可以使胰岛素的半衰期由4 min提高到 18 min。LEDGF(lens epithelium-derived growth factor)/p75是 HIV-1整合酶。Zvi等[23]通过对LEDGF/p75结构和功能的研究,取该蛋白质中的一段肽链,合成HIV-1整合酶抑制剂LEDPG 361-370,合成过程中应用Alloc保护的氨基酸,并进行特异性保护基的脱除,实现直链肽的首尾成环(图3)。LEDPG 361-370半衰期可长达8 d。

图3 LEDPG 361-370结构
4 氨基酸替换
替换肽链中的某几个氨基酸是另一种推迟酶降解使多肽药物的半衰期延长的方式,替换对象通常为肽链中的易酶解的氨基酸。
将L型氨基酸替换为D型非天然氨基酸是氨基酸替换的一种常规方法。西曲瑞克(cetrorelix)是一种促黄体激素释放激素(luteinizing hormone releasing hormone,LHRH)拮抗剂,用于治疗子宫纤维瘤、子宫内膜异位、卵巢癌以及改善良性前列腺肥大。西曲瑞克是将LHRH中的几个氨基酸替换为非天然的D型氨基酸,结构对比如图4所示[24]。西曲瑞克在多种酶存在的情况下半衰期可长达50 h,而LHRH的半衰期只有几小时[25]。从西曲瑞克的用法和剂量也可以看出其长效性:单剂量给药0.25 mg,作用可持续24 h;一次注射3 mg,药效可维持4 d。

图4 LHRH与西曲瑞克结构比较
Strausberg等[26]对枯草杆菌蛋白酶中随机的一段12个氨基酸组成的多肽链进行氨基酸替换,以对温度的稳定性和催化活性为基础进行筛选。筛选结果表明,在高温环境下,修饰后的产物半衰期最大可高达原枯草杆菌蛋白酶的15000倍。粒细胞多肽(human neutrophil peptides,HNP)是一种具有抗微生物活性和细胞毒性的多肽,其对胃癌细胞具有一定的毒性作用[27]。Linda 等[28]将人类嗜中性粒细胞肽(human neutrophil peptides,HNPs)序列中的精氨酸(Arginine)替换为鸟氨酸(Ornithine),通过药物代谢动力学研究发现168 h后仍有26%的经修饰后的HNP-Ornithine存在。
5 糖基化修饰
糖肽是指寡糖结构与多肽链中某些特殊氨基酸侧链上的功能团以共价键的形式相连接的一类多肽类物质[29]。糖基化修饰是指多肽附加上糖类的这一过程。在多肽的固相合成中,最常用的为N-糖基化和O-糖基化。N-糖基化通过天冬酰胺使侧链的酰胺氮进行连接。O-糖基化是指多肽与丝氨酸或苏氨酸残基上的氧相连接[30-31]。
多肽药物的糖基化修饰增加了侧链的空间位阻,提高了多肽对酶的稳定性。如alglucosidase alfa(myozyme,shire)用于治疗庞培氏病,按20 mg/kg(体质量)剂量,每2周注射1次[32]。darbepoetin alfa(aranesp,amgen)是促红细胞生长素(erythropoietin,EPO)糖基化修饰后的产品,用于治疗慢性肾功能衰竭及贫血,使用频率从老一代EPO的每周2~3次降低至每周1次或每2周1次[33]。TY027是一种阿片受体拮抗剂,在大鼠血浆中的半衰期只有4.8 h。为了得到更加稳定的止痛剂,Takashi等[34]以TY027为原型进行糖基化修饰,引入一个 O-β-(Ser(Glc)),以 Fmoc-Ser(O-β-D-Glc(OAc)4)-OH为原料,通过Fmoc固相合成多肽法得到TY027糖基化的修饰产物,TY027及该修饰后产物结构如图5所示。通过代谢稳定性实验,在大鼠血浆中孵化24 h仍然有70%完整的该修饰产物能被检测到。G.E.Umpierrez等[35]报道了LY2189265是一种新型的长效胰高血糖素(GLP-1)受体拮抗剂,用于治疗Ⅱ型糖尿病。通过糖基化修饰,该药物只需每周注射1次,大大提高了病人的依从性。

图5 TY027及糖基化后产物结构比较
6 PEG修饰
聚乙二醇(polyethyleneglycol,PEG)是由环氧乙烷聚合而成的大分子聚合物,结构式为HO-C(CH2CH2O)n-CH2CH2-OH。PEG类修饰剂具有无毒、良好的溶解性和生物相容性、宽泛的相对分子量选择范围等优点。经PEG修饰后的多肽类药物相对分子量有所增加,可降低肾小球的滤过效率,增强药物对酶的稳定性,从而延长药物在体内的半衰期[36]。
目前应用最普遍的PEG修饰剂是单甲氧基聚乙二醇(mPEG),结构为 CH3O-(CH2CH2O)n-CH2CH2-OH[37]。PEG修饰多肽类药物时,为了使PEG能在较温和的反应条件下与多肽偶联,故需先将PEG活化,再将活化后的PEG与多肽肽链的C端、N端以及侧链上的氨基、巯基和羧基进行偶联[38]。
1990年,美国食品与药品管理局(FDA)批准了第1个PEG修饰蛋白质类药物pegademase(adagen,enzon)上市。pegademase是由聚乙二醇修饰的腺苷脱氨酶,用于治疗严重的合并性免疫缺陷病症,给药频率为每周1次,从给药频率上体现了修饰后药物的长效性,减少了病人因频繁给药而带来的痛苦[39]。到目前为止,已有多种经PEG修饰的蛋白质多肽类药物获FDA批准上市销售,包括PEG修饰的天冬酰胺酶、重组人粒细胞集落刺激因子、干扰素等(表2)[40]。这些药物在临床应用中也体现出了PEG修饰的优势。从给药频率不难看出PEG修饰在延长蛋白质多肽类药物的半衰期上有显著作用。目前还有大批的研究者致力于PEG修饰多肽类药物的研究,也有很多新的PEG修饰药物处于研发甚至临床阶段[41-43]。但PEG修饰剂本身的分散性造成了修饰后的药物具有一定分散性,这会直接影响到产品的生物相容性、化学性能以及临床功效等,这也是目前困扰PEG修饰的一大难点。解决途径:①注重PEG的合成和纯化,以保证PEG修饰剂本身的质量;②特异性定点修饰技术的应用以及对最佳反应条件的探索。

表2 FDA批准上市的PEG修饰蛋白多肽类药物
7 结束语
据2010年调查,目前全世界市场销售的多肽类药物主要有51个品种,其中有6个品种在2008年销售额超过了7.5亿美元,总销售额高达88.3亿美元。但由于多肽在体内具有不稳定性、易被水解、易被肾脏清除等特性,使得多肽类药物半衰期短,从而必须频繁给药,因此对多肽药物的长效化研究显得尤为迫切。化学修饰是多肽类药物长效化技术中的常用方式,是药物设计的重要手段。在目前的临床应用中,通过化学修饰得到的长效多肽类药物已显现出其优良的性能。
在今后的研究中:一是对于药物靶向性的研究,通过特异性的化学修饰实现药物和病灶位点的特异性结合,使药物在发挥更好药效的同时,尽量降低对其他健康组织器官的伤害;二是将化学与生物方法相结合应用于多肽药物长效化技术的探索,集两者的优势,共同开发新型的复合型长效化技术;三是寻找便于实现大规模工业化生产的化学修饰方法,使药物从实验室研究走向工业化生产及上市销售,最终真正造福病人。
[1]Brian L B.Large-scale manufacture of peptide therapeutics by chemical synthesis[J].Nature Review,2003,2:587-593.
[2]黄宝斌,袁力勇,王军志.长效蛋白和多肽类药物的研发现状[J].国际生物制品学杂质,2008,31(1):31-34.
[3]Werle M,Bernkop-Schnurch A.Strategies to improve plasma half life time of peptide and protein drugs[J].AminoAcids,2006,30:351-367.
[4]Sabrina Trussel,Christoph Dumelin,Katharina Frey,et al.New Strategy for the Extension of the serum Half-life of Antibody Fragments[J].Bioconjugate Chem,2009,20:2286-2292.
[5]Qingyu Sun,Hosea Nelson,Tony Ly,et al.Side Chain Chemistry Mediates Backbone Fragmentation in Hydrogen Deficient Peptide Radicals[J].Journal of Proteome Research,2009,8:958-996.
[6]Sheng Jiang,Zheng Li,Ke Ding,et al.Recent Progress of Synthetic Studies to Peptide and Peptidomimetic Cyclization[J].CurrentOrganic Chemistry,2008,12:1502-1542.
[7]Samuel Blanquart,Nicolas Lartillot.A Site-and Time-Heterogeneous Model of Amino Acid Replacement[J].Mol Biol Evol,2008,25(5):842-858.
[8]Ricaedo J S,Kai G.Effects of Glycosylation on the Stability of Protein Pharmaceuticals[J].Journal of Pharmaceutical Sciences,2009,98(4):1223-1245.
[9]Roberts M J,Bentley M D,Harris J M.Chemistry for peptide and protein PEGylation[J].Advanced Drug Delivery Reviews,2002,54:459-476.
[10]Oyston P C F,Fox M A,Richardsand S J,et al.Novel peptide therapeutics for treatment of infections[J].Journal of Medical Microbiology,2009,58,977-987.
[11]Brinckerhoff L H,Kalashnikov V,Thompson L W,et al.Terminal modifications inhibit proteolytic degradation of an immunogenic MART-I(27-35)peptide:implications for peptide vaccines[J].Int J Cancer,1999,83:326-334.
[12]Dasgupta P,Singh A,Mukherjee R.N-terminal acylation of somatostatin analog with long chain fatty acids enhances its stability and anti-proliferative activity in human breast adenocarcinoma cells[J].Biol Pharm Bull,2002,25:29-36.
[13]付慧君,周宁,周英等.抗蛋白酶降解肽类药物的结构修饰研究进展[J].国外医药:医学分册,2006,33(4):269-271.
[14]Sabrina Trussel,Christoph Dumelin,Katharina Frey,et al.New Strategy for the Extension of the Serum Half-Life of Antibody Fragments[J].Bioconjugate Chem,2009(20):2286-2292.
[15]David Sabatino,Caroline Proulx,Sophie Klocek,et al.Exploring Side-Chain Diversity by Submonomer Solid-Phase Aza-Peptide Synthesi s[J].Organic Letters,2009,11(16):3650-3653.
[16]Norbert Sewald,Hans-Diter Jakubke.Peptide:Chemisty and biology[M].Germany:Wiley-VCH Verlag Publ,2002:135-256.
[17]Dong Xie,He Jiang.An Albumin-Conjugated Peptide Exhibits Potent Anti-Human Immunodeficiency Virus Activity and Long In Vivo Half-Life[J].ANTIMICROBIAL AGENTS AND CHEMOTHERAPY,2010,54(1):1-6.
[18]Peng Wu,Wenqing Shui,Brian L.Carlson,et al.Site-specific chemical modification of recombinant Proteins produced in mammalian cells by using the genetically encoded aldehyde tag[J].PANS,2009,106(9):3000-3005.
[19]王德心,韩香,龚喜等.环肽的合成研究进展[J].有机化学,2008,28(4):549-573.
[20]Soledad R G,Norbert Sewald.Synthesis of chemically modified bioactive peptide:recent advances,challenges and developments for medicinal chemistry[J].Future Med Chem,2009,1(7):1289-1310.
[21]Su C M,J ensen L R,Heimer E P,et al.In vitro stability of growth hormone releasing factor(GRF)analogs in porcine plasma[J].Horm Metab Res,1991,23:15-21.
[22]Rozek A,Powers JP,FriedrichCL,et al.Structure-based design of anindolicidin peptide analogue with increased protease stability[J].Biochemistry,2003,42:14130-14138.
[23]Zvi Hayouka,Mattan Hurevich,Aviad Levin,et al.Cyclic peptide inhibitors of HIV-1 integrase derived from the LEDGF/p75 protein[J].Bioorganic & Medicinal Chemistry,2010,18:8388-8395.
[24]蔡佳利,陈朝晖,耿蓉霞,等.抗癌药西曲瑞克应用研究新进展[J].中国实用医药,2008,3(3):1-3.
[25]Reissmann T,Engel J.The LHRH antagonist cetrorelix[J].Drugs Future,1994,19:228-237.
[26]Strausberg S L,Ruan B,Fisher K E,et al.Directed coevolution of stability and catalytic activity in calcium-free subtilisin[J].Biochemistry,2005,44:3272-3279.
[27]刘文超,张学庸,胡家露.人中性粒细胞多肽体外杀伤肿瘤细胞的实验研究[J].第四军医大学学报,1999,20(9):408-411.
[28]Linda A S,Rodney L L,Bernadette R,et al.ADP-ribosylation of human defensin HNP-1 results in the replacement of the modified arginine with the noncoded amino acid ornithine[J].PNAS,2009,106(47):19796-19800.
[29]张法,刘刚.糖肽的固相合成[J].化学进展,2006,18(5):579-600.
[30]Wei X,Li L.Comparative glycoproteomics:approaches and application[J].Brief Funct Genomic Roteomic,2008,8(2):104-113.
[31]Ricardo J S,Griebenow K.Glycosylation of Therapeutic Proteins:An Effective Strategy to Optimize Efficacy[J].Bio Drugs,2010,24(1):9-21.
[32]Clark S E,Muslin E H,Henson C A.Effect of adding and removing N-glycosylation recognition sites on the thermostability of barley alpha-glucosidase[J].Protein Eng Des Sel,2004,17(3):245-249.
[33]Marc A P,Emmanuel A B,Chao-Yin Chen,et al.Baseline Characteristics in the Trial to Reduce Cardiovascular Events With Aranesp Therapy(TREAT)[J].A-merican Journal of Kidney Diseases,2009,54(1):59-69.
[34]Takashi Yamamoto,Padma Nair,NeilE Jacobsen,et al.Improving Metabolic Stability by Glycosylation:Bifunctional Peptide Derivatives That Are Opioid Receptor Agonists and Neurokin in 1 Receptor Antagonists[J].J Med Chem,2009,52:5164-5175.
[35]Umpierrez G E,Blevins T,Rosenstock J,et al.The effects of LY2189265,a long-acting glucagon-like peptide-1 analogue,in a randomized,placebo-controlled,double-blind study of overweight/obese patients with Type 2 diabetes:the EGO study[J].Diabetes,Obesity and Metabolism,2011,13:418-425.
[36]边蕾,石屹峰.蛋白质药物长效化技术的现状和进展[J].中国生物工程杂志,2009,29(1):114-118.
[37]Roberts M J,Bentley M D,Harris J M.Chemistry for peptide and protein PEGylation[J].Advanced Drug Delivery Reviews,2002,54:459-476.
[38]路娟,罗国安,王义明,等.药物的聚乙二醇修饰研究进展[J].有机化学,2009,29(8):1167-1174.
[39]Hershfeld M S.Adenosine deaminase deficiency:clinical expression,molecular basis,and therapy[J].Semin Hematol,1998,35:291-298.
[40]Jung Seok Kang,Patrick P DeLuca,Kang Choon Lee.E-merging PEGylated drugs[J].Expert Opin,2009,14(2):363-380.
[41]Youn Y S,Chae S Y,Lee S,et al.Evaluation of therapeutic potentials of site-specific PEGylated glucagons-like peptide-1 isomers as a type 2 anti-diabetic treatment:insulinotropic activity,glucose-stabilizing capability,and proteolytic stability[J].Biochem Pharmacol,2007,73:84-93.
[42]Youn Y S,Lee K C.Site-speciic PEGylation for highyield preparation of Lys21-amine PEGylated growth hormone-releasing factor(GRF)(1-29)using a GFR(1-29)derivative FMOC-protected at Tyr1 and Lys12[J].Bioconjug Chem,2007,18:500-506.
[43]Youn Y S,Na D H,Lee K C.High-yield production of biologically active mono-PEGylated salmon calcitonin by site-speciic PEGylation[J].J Control Rel,2007,117:371-379.
猜你喜欢
山东农业大学学报(自然科学版)(2021年3期)2021-07-29
中国计算机报(2019年26期)2019-08-27
世界农药(2019年2期)2019-07-13
中学生物学(2019年2期)2019-04-16
中学课程辅导·教师通讯(2018年6期)2018-06-20
粘接(2017年4期)2017-04-25
中学生数理化·高二版(2017年2期)2017-04-19
中学化学(2016年12期)2017-02-05
物理化学学报(2015年7期)2015-12-30
原子与分子物理学报(2015年3期)2015-11-24
