
Khimera软件在Hg氯化动力学机理研究中的应用
2012-09-14 03:13:58柏静儒杨博文
东北电力大学学报 2012年6期
柏静儒,杨博文,王 擎
(东北电力大学能源与动力工程学院,吉林 吉林 132012)
固体燃料利用过程中,痕量元素的迁移及形态转化是一个复杂的过程,研究表明痕量元素的毒性依赖于元素的物理化学形式以及浓度,不同形态元素间的毒性以及环境行为有较大的区别。了解燃煤烟气中痕量元素的形态对认识其迁徙富集规律、排放的抑制以及评估其对人类健康和环境的危害等问题有重要的意义,因此测定和预报燃煤烟气中痕量元素的形态分布以及浓度变化十分重要。化学反应动力学研究是确定化合物反应机理并最终确定元素形态以及浓度和时间依变关系的一种可靠方法,但必须基于对痕量元素不同组分的总包与基元反应的动力学参数的完整性和精确性[1],特别是各化学反应的反应速率常数的精确获得,是煤等燃料燃烧过程痕量元素动力学研究的核心问题。
Widmer提出了较为完整的汞的氧化动力学模型,但其基元反应的动力学参数还是相当粗糙的。乔瑜等[2]使用CHEMKIN软件包对Hg/O/H/Cl系统中汞的氧化动力学进行了研究,每个基元反应的动力学参数取自其它文献及其试验数据,其计算计算结果与Mamani-Paco等人的试验数据间的误差可能是由于汞的氧化动力学参数的偏差。王臣等[3]采用经典过渡态理论计算了燃煤烟气中Hg的三个基元的反应速率常数,发现其计算结果同Widmer得到的速率常数之间存在一定的差异,并认为这种差异主要是由于Widmer在利用Glar-borg的燃烧化学机理推导反应速率常数的过程存在一定得错误。可见,对燃烧中有痕量元素的生成与排放的动力学模拟而言,基元反应的反应速率常数是关键的输入参数。而量子化学从头计算理论的发展为反应速率常数等动力学参数的计算提供了可能,基于量子化学从头计算理论,很多研究者对煤燃烧烟气中痕量元素的反应的微观机制进行研究,并对动力学参数进行求解。刘晶等[4]量子化学从头计算MP2方法,在SDD基组水平上研究了燃烧过程中HgCl氯化反应以及汞与N2O和O3反应的微观机理,采用经典过渡态理论计算各反应在T=1000 K时的动力学参数。Niksa等[5,6]采用修正过的Hg的反应动力学模型,研究了烟气成分对汞的反应的影响。可见,Hg的反应均是利用量化理论对痕量元素所发生反应的机理进行了研究和分析,并也证明了量子化学从头计算在研究燃烧过程痕量元素反应动力学上的可行性,反应速率常数仍按经典过渡态理论的公式来计算,其结果都是在特定的温度下的某一个特定的值,而缺少反应速率常数同反应温度之间的依变关系。而基元反应速率常数对于燃烧过程中痕量元素的生成和排放的动力学模拟起着举足轻重的作用,因此有必要进行精确分析,获得更为准确的动力学参数值,以发展和完善燃烧过程中痕量元素的反应动力学模型,才能很好的描述痕量元素的反应产物及其浓度与时间的关系。
本文借助量子化学软件Khimera,选取有关痕量元素Hg的两个典型基元反应,在温度区间[200 K,2000 K]进行研究,为全面建立Hg的化学反应动力学模型奠定基础。
1 研究方案与方法
本部分选取的涉及痕量元素Hg的两个基元反应(1)和(2)进行研究。

首先采用量子化学软件Gaussian03[7]的配套软件Gaussview,对两个基元反应(1)和(2)中所涉及的分子进行初始构型。然后应用Gaussian03软件对所构造的分子进行优化,进而在相同水平上计算能量、频率等参数。最后计算并优化反应过渡态,同时得到其相应的频率、能量值,进而确定反应通道。上述计算过程中采用量子化学从头计算中电子二级微扰理论MP2方法,考虑到Hg是大质量多电荷的金属元素,对其采用SDD基组,其余元素都采用6-311++G(3df,3pd)基组。
在上述研究的基础上,用Khimera软件对反应(1)和(2)在温度区间[200 K,2000 K]分别进行反应速率常数计算。
2 Khimera软件
Khimera是Kintech公司和摩托罗拉Digital DNA实验室(现为Freescale)合作研发的一款非常独特的工具软件,主要用于从量子化学计算的结果中提取数据并做进一步处理,从而获得微观反应过程的热/动力学性质、基元反应速率和物质的传导性等参数。Khimera帮助工作在燃烧、等离子体化学和材料科学等领域的科研工作者应用量子电子学和量子化学软件来进行原子模拟的工具。它是研究和分析化学过程的软件包,它简便反应机理发展,从基本的分子数据中估测热力学、传送和动力学参数,进行反应器水平的模拟。目前Khimera的国内用户较少。
2.1 主要特点
Khimera可以处理气体、等离子体以及气-固界面反应中涉及到的复杂的物理化学变化过程,目前几乎没有其它商业软件能够提供类似的功能。该软件的主要特点为:动力学机理建模包括敏感度分析和自动的机制缩减能力;使用多种现代理论方法来计算基本的单分子和双分子反应的速率常数;直接从著名的量子化学代码Gaussian,ADF,Jaguar,GAMESS的输出文件中导入数据;含有大量物质、反应和化学机理的动力学和热力学参数的数据库;一系列的反应器模块可以模拟所有的反应过程,并且可以证明/分析反应机理。同时Khimera包含一个准-3D分子察看器,以图形方式显示计算结果。Khimera有文件导入/导出的能力,可以方便的应用在各种反应机理研究和动力学分析中。
2.2 功 能
Khimera可识别量子化学计算的结果(目前支持Gaussian,GAMESS和Jaguar程序),这些计算结果都是关于基本的气-气和气-液反应,而这些反应都包含所研究化学过程的(假设)机理。这些结果(能量,频率,以及反应物,过渡态和最终产品的结构)被用来计算焓值,熵值和单个反应的速率。
(1)过渡态理论和直接生物分子反应;
(2)带有或不带有强碰撞假设的单分子RRKM模型;
(3)经历长寿命中间体络合物的分子气相反应模型;
(4)基于普遍过渡态理论和统计理论,并考虑到表面扩散的微观表面动力学模型;
(5)同时依据Langevin方法和统计理论的离子-分子反应模型;
(6)反应速率参数和热动力性质的计算结果将被输出为CHEMKIN输入格式,以便进一步利用动力学软件包进行分析。
3 结果分析
3.1 微观反应历程分析
表1列出了分子构型优化的结果与实验值、文献[7]结果的比较。由表中数据可以得出,本文计算结果与美国国家标准技术研究所(NIST)实验值的误差分别为0.22%(HCl)、6.88%(HgCl)和1.60%(HgCl2)。文献的结果误差分别为:3.12%(HCl)、10.79%(HgCl)和1.68%(HgCl2),与之相比更为精确,与实验值更加吻合。

表1 分子几何构型优化结果的比较
所构建的Hg的两个基元反应(1)和(2)的微观反应机理如图1所示,采用量子化学从头计算理论MP2方法,基组选择SDD对反应通道上的过渡态进行优化。
由图1(a)可见,在Hg与HCl反应过程中,Hg原子靠近HCl中Cl原子的同时H-Cl键长逐渐拉长,此时Hg-Cl键将成未成H-Cl键将断未断而形成存在时间极短的过渡态{HgClH}#,随着H-Cl键继续拉长,H-Cl键断裂的同时Hg-Cl键形成从而反应最终完成。由图1(b)可见,HgCl+HCl=HgCl2+H反应过程中,HgCl与HCl分子中的Hg原子和Cl原子相互靠近,与此同时Hg-Cl键、H-Cl键逐渐拉长从而形成过渡态{ClHgClH}#,此时Hg-Cl键将成未成H-Cl键将断未断。伴随两分子继续靠近,H-Cl断开Hg-Cl形成,反应结束。
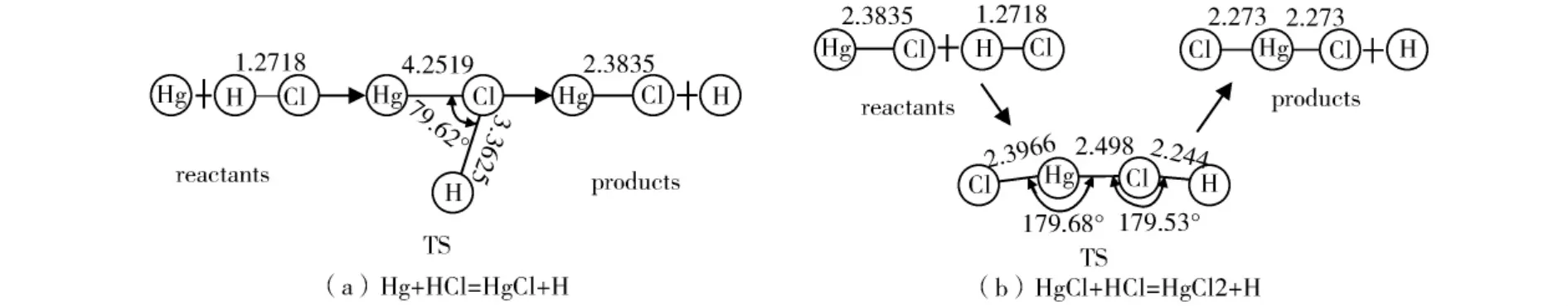
图1 Hg氯化反应微观历程
3.2 各基元反应速率常数分析
基于量子化学从头计算所构建的反应的微观历程,采用Khimera软件分别计算了两个基元反应的反应速率常数,温度区间[200 K,2000 K],结果见图2。
图2(a)为Hg+HCl=HgCl+H反应的k-T图,由图可知此反应为可逆反应,同一温度下正反应的速率常数大于逆反应的速率常数,即正反应比逆反应更容易进行。温度在200 K至500 K时,反应速率常数随温度的增加而迅速增加,但数值均较小下;温度在500 K至2000 K时,反应速率常数随温度的增加速度减缓,曲线较为平稳。经过拟合计算得出反应速率表达式如式(3)所示。图2(b)为HgCl+HCl=HgCl2+H反应的k-T图,从图中可以看出此反应只能单向进行,在温度小于500 K时,反应速率常数随温度的增加变化显著,在500 K以后变化逐渐趋于平稳。此反应的反应速率的表达式如式(4)所示。


图2 Hg氯化反应过程温度与反应速率常数关系图示
4 结论
(1)由几何构型与能量分析结果可知,采用Gaussian03软件对痕量元素进行研究的结果与实验结果吻合较好;
(2)反应速率常数的计算结果表明,随着温度的增加反应也随之加快。两个基元反应之中,Hg+HCl=HgCl+H是可逆反应,其同一温度下正反应速率大于逆反应,说明正反应更容易进行。温度在200 K至500 K时,反应速率常数随温度的增加变化显著,而500 K至2000 K时反应速率常数随着温度增加的变化趋势减弱,逐渐趋于平稳;
(3)本文研究结果表明,应用量子化学软件Khimera对Hg等有毒痕量金属元素基元反应的反应速率常数进行研究是有效可行的。
[1]郭欣,贾小红,郑楚光.燃煤烟气中痕量元素的形态分析方法进展[J].环境科学与技术,2005,28(1):100-102.
[2]乔瑜,徐明厚,冯荣,等.Hg/O/H/Cl系统中汞的氧化动力学研究[J].中国电机工程学报,2002,22(12):138-141.
[3]王臣,徐明厚,刘晶,等.基于量子化学的燃煤过程痕量元素反应机理研究及其进展[J].热力发电,2005,34(4):15-19.
[4]刘晶,郑楚光,徐明厚,等.燃烧中汞与 N2O、O3的反应机理[J].燃烧科学与技术,2005,11(2):155-158.
[5]Niksa S,Fujiwara N,Fujita Y,et al.A mechanism for mercury oxidation in coal-derived exhausts[J].Journal of the Air& Waste Management Association,2002,52(8):894 -901.
[6]Niksa S,Helble J,Fujiwara N.Kinetic modeling of homogenous mercury oxidation:the importance of NO and H2O in predicting oxidation in coal-derived systems[J].Environmental Science and Technology,2001,35(18):3701 -3706.
[7]Frisch M J,Trucks G W,Schlegel H B,et al.Gaussian 03,Revision B.01[S].Gaussian,Pittsburgh PA,2003.
[8]刘晶,郑楚光,邱建荣.燃烧烟气汞反应的量子化学计算方法研究[J].工程热物理学报,2007,28(03):519-521.
猜你喜欢
高中数理化(2023年6期)2023-08-26 13:28:24
黑龙江大学自然科学学报(2022年4期)2022-11-17 08:07:40
北京航空航天大学学报(2022年5期)2022-06-06 09:27:18
大学化学(2021年8期)2021-09-26 10:51:16
理化检验-化学分册(2020年5期)2020-06-15 11:36:04
科学导报(2018年30期)2018-05-14 12:06:01
电脑知识与技术(2018年3期)2018-03-21 09:27:04
哈尔滨理工大学学报(2017年1期)2017-04-08 04:16:24
西藏科技(2015年1期)2015-09-26 12:09:23
质谱学报(2015年5期)2015-03-01 03:18:47
