
PERK通路参与了缺氧心肌细胞的内质网应激及凋亡*
2012-09-07 03:31:40刘春蕾李蕊君何云云何昆仑王莉莉
中国病理生理杂志 2012年8期
刘春蕾, 李 鑫, 李蕊君, 何云云, 何昆仑△, 王莉莉
(1中国人民解放军总医院心内科,北京 100853;2南开大学医学院,天津 300071;3军事医学科学院毒物药物研究所,北京 100850)
PERK通路参与了缺氧心肌细胞的内质网应激及凋亡*
刘春蕾1,2, 李 鑫1, 李蕊君1, 何云云1,2, 何昆仑1△, 王莉莉3△
(1中国人民解放军总医院心内科,北京 100853;2南开大学医学院,天津 300071;3军事医学科学院毒物药物研究所,北京 100850)
目的:观察缺氧对原代培养的Wistar乳鼠心肌细胞的损伤,探讨内质网应激在缺氧心肌损伤发生发展过程中起的作用及PERK通路是否参与其信号转导过程。方法:将原代培养的乳鼠心肌细胞随机分为正常对照组和缺氧1 h、4 h、8 h、12 h、24 h组,通过测定细胞ATP含量反映细胞活力;高内涵分析细胞成像系统检测多参数凋亡;采用免疫细胞化学和蛋白印迹方法检测以内质网为靶点的分子伴侣(GRP78和钙网蛋白)的表达,PERK通路(PERK和eIF2α)的磷酸化水平,以及其下游分子(ATF4和CHOP)在缺氧不同时点蛋白的表达变化特征。采用PERK通路激活型药物salubrinal处理原代培养的Wistar乳鼠心肌细胞,观察药物是否对缺氧损伤的心肌细胞有保护作用。结果:缺氧引起心肌细胞凋亡,缺氧早期(约1 h)钙网蛋白和GPR78的表达上调;缺氧中期(4 h)p-PERK、p-eIF2α和ATF4的表达上调;缺氧后期(12 h)CHOP的表达上调。Salubrinal对缺氧心肌有保护作用。结论:在培养的心肌细胞中,缺氧可激发内质网应激。在缺氧早期激活PERK通路保护机体对抗缺氧损伤,后期激活细胞凋亡通路。
心肌细胞;内质网应激;缺氧;未折叠蛋白反应;细胞凋亡
心肌缺血触发心肌细胞发生持续性凋亡反应,使心肌细胞进行性丢失、胶原支架断裂、心室扩张和心室重塑,是多种心脏不良事件发生的重要原因[1]。因此,深入阐明缺氧心肌细胞凋亡的分子机制可望为缺血缺氧相关的心脏疾病提供新的有效干预手段。
传统的细胞凋亡的途径又称caspase依赖的细胞死亡途径,主要包括死亡受体途径和线粒体途径,两者均通过诱导caspase级联反应激活caspase-3,导致细胞凋亡。近年的研究显示,除了caspase依赖的细胞凋亡途径外,内质网应激(endoplasmic reticulum stress,ERS)也与细胞凋亡密切相关[2]。ERS是由于细胞内外环境的改变引起ER内未折叠蛋白质聚集,钙稳态失衡,从而导致的内质网功能紊乱的状态。在ERS早期,未折叠蛋白反应(unfolded protein response,UPR)是重要特征,具有保护细胞抵抗外界环境对细胞的干扰作用;然而,一旦ERS持续而严重,则激活凋亡通路而诱发细胞凋亡[3]。研究表明,UPR可由3条独立的信号通路所诱导,包括活化转录因子6(activating transcription factor 6,ATF6)、肌醇需酶(inositol-requiring enzyme 1,IRE1)和蛋白激酶R样内质网激酶(protein kinase R-like ER kinase,PERK)[4]。目前,在心肌细胞中,缺氧诱发细胞ERS以及凋亡已经得到广泛证实。Negoro等[5]和 Ron等[6]观察到,缺氧确能诱导新生乳鼠心肌细胞产生UPR反应。也有研究发现,心肌细胞在缺氧条件下,ATF6通路蛋白表达上调[7],诱发ATF6和IRE1通路共同的下游分子X盒结合蛋白1(X-box-binding protein 1,XBP1)及凋亡蛋白的表达,并随着缺氧时间延长,最终使细胞死亡[8]。由此可见,ATF6和IRE1通路参与了缺氧心肌细胞ERS及凋亡。相比较ATF6和IRE1通路而言,PERK抑制蛋白合成途径,更容易通过增强核糖体真核生物起始因子2(eukaryotic initiation factor 2,eIF2α)磷酸化酶的活性或抑制其去磷酸化酶的活性,抑制细胞凋亡。然而,PERK通路是否也参与了心肌细胞缺氧过程尚不清楚。本实验通过缺氧诱导原代培养乳鼠心肌细胞凋亡,采用免疫细胞化学和蛋白印迹的方法,观察了缺氧不同时间对内质网应激及凋亡相关蛋白表达的影响,特别是PERK通路相关蛋白的变化特点,旨在揭示PERK信号通路与缺氧诱导心肌细胞损伤的关系。
材料和方法
1 材料
新生24 h内的Wistar大鼠,雌雄不拘(由军事医学科学院实验动物中心提供),动物合格证号为SCXK-(军)2007-004。小鼠抗大鼠GAPDH单克隆抗体、兔抗大鼠葡萄糖调节蛋白质78(glucoseregulated protein,GRP78)多克隆抗体购自Stressgen。兔抗大鼠钙网蛋白(calreticulin,Cal)多克隆抗体、兔抗大鼠phospho-PERK多克隆抗体、兔抗大鼠PERK多克隆抗体、兔抗大鼠eIF2α多克隆抗体、兔抗大鼠phospho-eIF2α多克隆抗体和小鼠抗大鼠C/EBP同源蛋白质(C/EBP homologous protein,CHOP)多克隆抗体购自Cell Signaling Technology。兔抗大鼠活化转录因子4(activating transcription factor 4,ATF4)多克隆抗体购自Abcam。山羊抗小鼠IgG DylightTM594购自 Thermo scientific。ATP试剂盒购自 Promega。Salubrinal(Sal)和Hoechst 33258购自Invitrogen。高内涵分析系统 In cell Analyzer 2000为 GE产品; Western blotting成像与分析采用的Alphamager 5500凝胶成像系统为Alpha产品。
2 方法
2.1 心肌细胞原代培养 迅速取出新生Wistar大鼠心脏,剪碎,用终浓度为0.125%胰蛋白酶和0.1%胶原酶在37℃水浴中消化20 min,用含10%FBS的DMEM培养基中和蛋白酶,将细胞悬液离心并重悬细胞接种至75 cm2培养瓶或培养板中,37℃、5%CO2孵箱培养24 h后半量换液;之后每48 h换液1次。4 d后,缺氧处理的细胞置于密闭的恒温缺氧仓内,通入95%N2/5%CO2的混合气体,O2含量少于0.5%。
2.2 细胞活力测定 纯化的乳鼠心肌细胞按1× 104cells/well接种到96孔板37℃、5%CO2孵箱培养4 d,之后置缺氧箱中分别缺氧处理细胞1 h、4 h、8 h、12 h、24 h,每时点设3个重复孔。同时设置药物干预组,细胞缺氧处理前 24 h使用不同浓度的PERK通路激活型药物salubrinal孵育心肌细胞30 min,按照细胞活力检测试剂盒说明检测细胞活性。采用酶标仪检测562 nm波长下的荧光值。
2.3 多参数凋亡检测试剂盒检测细胞凋亡[9]每孔加入10 000个细胞,在37℃、95%N2、5%CO2的缺氧箱中孵育36 h,加入5%线粒体膜电位和DNA荧光素染料MitoTracker/Hoechst溶液,室温染色10 min,PBS清洗后,细胞进行透化处理,鬼笔环肽(Alexa Fluor 488 phalloidin solution)室温暗处理30 min。剩余的溶液用PBS洗3次。应用特异性的DNA荧光素染料 Hoechst 33258,线粒体膜电位染料 Mito-Tracker,细胞骨架染料Alexa Fluor 488进行染色后,用高内涵分析细胞成像系统在波长350/461 nm、495/519 nm和579/599 nm下进行检测观察。检测内容包括细胞形态、细胞核面积、DNA含量、核强度、肌动蛋白含量、线粒体膜电位等指标。正常细胞核为圆形或椭圆形,荧光着色浅,凋亡细胞核固缩,呈强荧光反应;与正常细胞相比,凋亡细胞骨架微丝蛋白含量降低,线粒体膜电位下降等现象
2.4 Western blotting法检测心肌细胞蛋白表达 纯化的乳鼠心肌细胞按1×106cells/well接种到6孔板,在37℃、5%CO2孵箱培养4 d后,在95%N2、5%CO2的缺氧箱中孵育不同时间。用1%SDS裂解液(mmol/L:NaCl 100,EDTA 0.1,DTT 10,HEPES 50)处理心肌细胞,离心取上清,用BCA法进行蛋白定量。于100℃水浴加热10 min,12 000×g、4℃离心10 min。经10%SDS-PAGE电泳分离后,将目标条带电转至PVDF膜,其中PERK和p-PERK用25 V半干式转移30 min,其余采用15 V半干式转移15 min;之后,采用5%脱脂奶粉室温封闭电转后的PVDF膜1 h,Ⅰ抗分别为GRP78(1∶1 000)、calreticulin(1∶200)、PERK(1∶1 000)、p-PERK(1∶1 000)、phospho-eIF2α(1∶1 000)、eIF2α(1∶1 000)、ATF4(1∶500)、GAPDH(1∶200)、β-actin(1∶500),4℃孵育过夜,TBS/T洗3次,加入Ⅱ抗(1∶5 000)室温孵育1 h,TBS/T洗3次;之后加SuperECL Plus超敏发光液避光孵育 3 min,最后采用 AlphaImager 5500凝胶成像系统进行成像拍摄。用 FluorChem 5500进行蛋白量灰度分析。
2.5 免疫细胞化学方法检测CHOP蛋白表达 纯化的乳鼠心肌细胞按1×104cells/well接种到黑色底透96孔板,在37℃、5%CO2孵箱培养4d后,置于37℃、95%N2和5%CO2缺氧箱中孵育不同时间。之后,细胞用4%多聚甲醛固定15 min。封闭液室温封闭1 h;1∶1 600稀释Ⅰ抗CHOP,4℃孵育过夜; PBS洗3次,随后用含Hoechest 33258(3 μmol/L)的荧光Ⅱ抗(1∶500)37℃孵育2 h。洗后,用In Cell Analyzer 1000高内涵分析细胞成像系统检测,采用In Cell Analyzer Workstation进行荧光强度分析。
3 统计学处理
数据以均数±标准差(珋x±s)表示,采用SPSS 13.0统计软件,组间比较采用单因素方差分析(One way-ANOVA),以P<0.05为差异有统计学意义。
结果
1 缺氧对大鼠心肌细胞ATP含量的影响
细胞ATP含量测定表明,与正常对照组相比,缺氧1 h、4 h、8 h、12 h、24 h后,细胞ATP含量显著下降(P<0.05),随着缺氧时间的延长,细胞ATP含量呈减低趋势,但缺氧4 h较缺氧1 h有一升高,见图1。
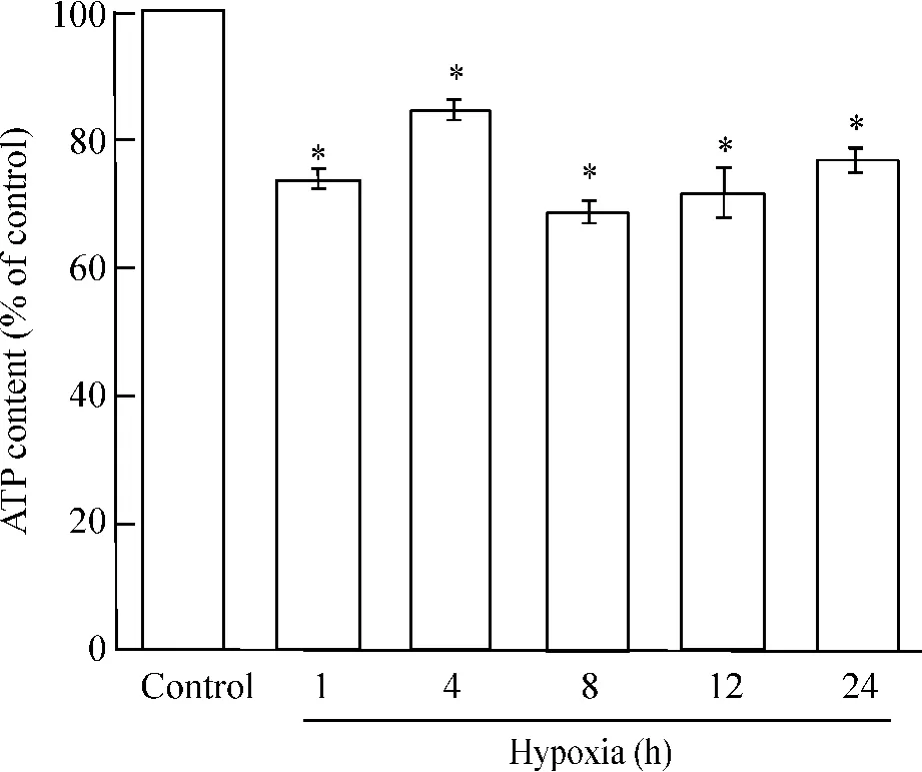
Figure 1. The effect of hypoxia on the viability of primary neonatal rat cardiomyocytes at different time points.x珋±s.n=3.*P<0.05 vs control(non-hypoxia treatment) group.图1 缺氧不同时点对原代培养新生大鼠心肌细胞ATP含量的影响
2 凋亡心肌细胞的鉴定
多参数凋亡检测试剂盒检测细胞凋亡的结果显示:缺氧后,细胞出现核固缩、骨架微丝蛋白含量下降、线粒体膜电位下降等凋亡细胞的特征,见图2。从这3个参数的数据分析显示缺氧16 h和24 h造成明显的细胞凋亡,核荧光强度分别较对照组高26.3%和26.6%;细胞骨架微丝含量分别较对照组低5%和11%;线粒体膜电位分别较对照组低17%和27%(P<0.05)。
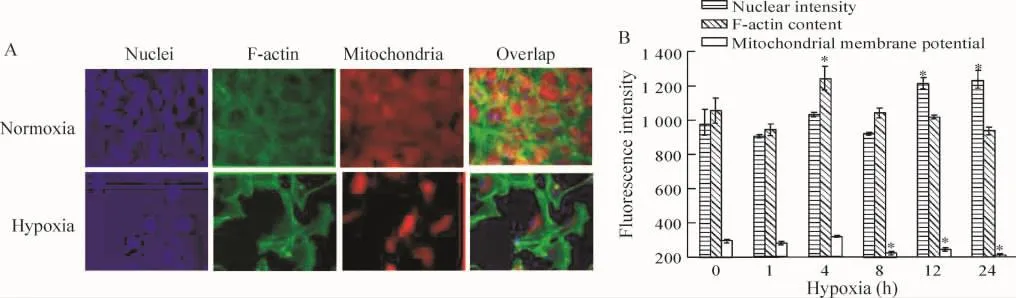
Figure 2. The effect of hypoxia on the apoptosis of the cardiomyocytes detected by high-content analysis.A:the images with nuclear staining,F-actin staining and mitochondrial membrane potential staining under normoxia or hypoxia for 24 h(×400);B: the nuclear intensity,F-actin content and mitochondrial membrane potential were statistical analyzed.珋x±s.n=3.*P<0.05 vs control group(0 h).图2 多参数检测缺氧不同时间后心肌细胞的凋亡情况
3 Sal对缺氧诱导心肌细胞ATP含量的影响
细胞ATP含量测定发现,提前30 min给予心肌细胞salubrinal 10、20、40 μmol/L,再缺氧培养24 h后,与单纯缺氧组相比,salubrinal预给后增加细胞活力,细胞ATP含量显著上升,见图3。
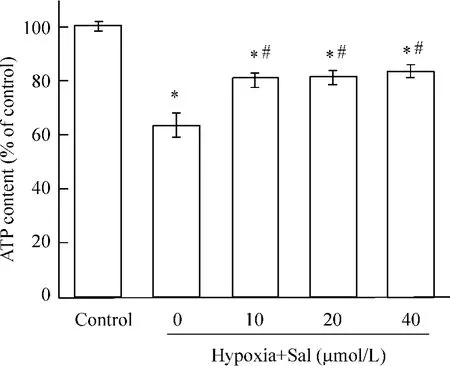
Figure 3. The protective effect of salubrinal(Sal)on hypoxic cardiomyocytes±s.n=3.#P<0.05 vs hypoxia+ Sal(0 μmol/L)group;*P<0.05 vs control group.图3 不同浓度salubrinal对缺氧心肌细胞的保护作用
4 缺氧对心肌细胞内质网应激相关因子的影响
4.1 缺氧对心肌细胞内质网应激标志分子(钙网蛋白和GRP78)表达的影响 与正常对照相比,缺氧1 h,心肌细胞内质网应激激活蛋白GRP78、钙网蛋白显著增加,其中钙网蛋白较对照组高72%(P<0.05),见图 4A,GRP78较对照组高 65%(P<0.05),见图4B;但随着缺氧时间的延长,钙网蛋白在缺氧4、8和12 h与对照组表达量相当,缺氧24 h显著低于对照组;而GRP78在缺氧4、8 h与对照组表达量相当,缺氧12 h、24 h显著低于对照组。上述结果表明,心肌细胞内质网应激分子钙网蛋白和GRP78表达均呈现早期升高,后期下降的趋势。
4.2 缺氧对PERK和eIF2α磷酸化水平及ATF4蛋白表达的影响 Western blotting测定结果显示,与正常对照组相比,从缺氧 1 h起,心肌细胞 p-PERK/PERK开始显著升高,至缺氧4 h,p-PERK/ PERK达到最大,较对照组高73%,随后逐渐下降,见图5A,缺氧8 h仍保持较高水平,但到12 h后则低于对照水平;缺氧心肌细胞eIF2α的磷酸化水平也是从缺氧1 h起开始显著升高,至缺氧4 h,peIF2α/eIF2α达到最大,较对照组高87%(P<0.05),见图5B,随后虽逐渐下降,但直至缺氧24 h仍然保持较高表达水平;对照组ATF4不表达,缺氧后,细胞中ATF4蛋白表达量升高,在4 h达到最高峰,随后显著下降(P<0.05),见图5C,但至缺氧24 h仍有表达。
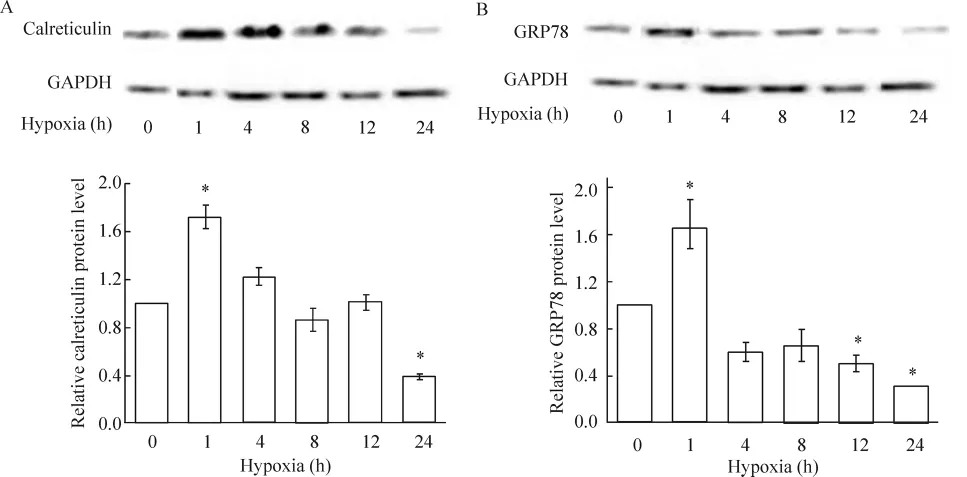
Figure 4. Effect of hypoxia on calreticulin(A)and GRP78(B)protein expression.珋x±s.n=3.*P<0.05 vs control(0 h)group.图4 缺氧不同时间对钙网蛋白和GRP78蛋白表达的影响
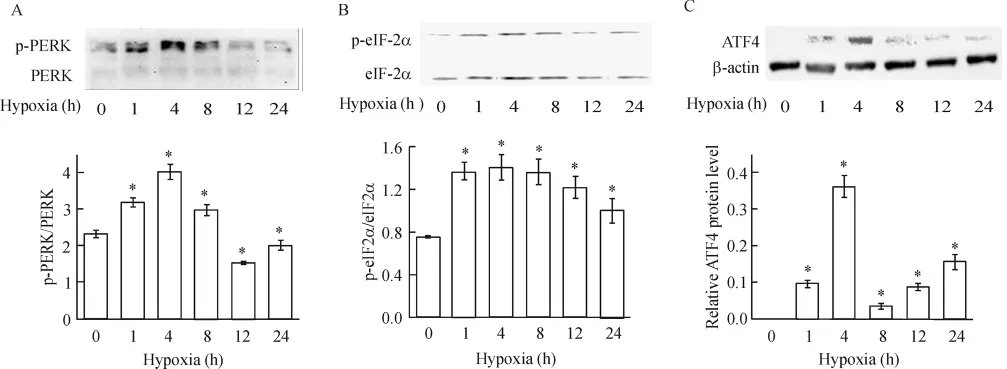
Figure 5. Effect of hypoxia on p-PERK/PERK(A),p-eIF2α/eIF2α(B)and ATF4(C)protein expression.珋x±s.n=3.*P<0.05 vs control(0 h)group.图5 缺氧不同时间对p-PERK/PERK、p-eIF2α/eIF2α及ATF4蛋白表达的影响
4.3 缺氧对内质网应激特异性凋亡分子(CHOP)表达的影响 由免疫细胞化学结果可以看出,内质网应激特异性凋亡分子CHOP在非缺氧处理的对照组和短期缺氧组心肌细胞上不表达,但在缺氧12 h和 24 h,CHOP出现表达。可见,缺氧可激活PERK通路下游的凋亡活性分子CHOP,缺氧12 h心肌细胞凋亡相关蛋白达到较高水平(P<0.05),见图6。
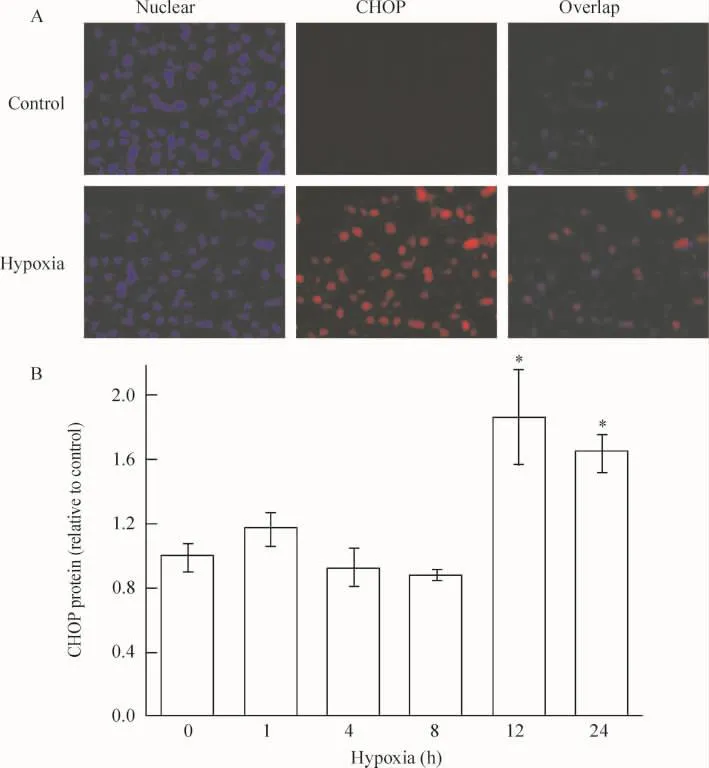
Figure 6. Effect of hypoxia on CHOP protein expression.A:the representative images of high-content analysis(×200);B:the fluorescence intensity analysis of CHOP.珋x±s.n=3.*P<0.05 vs control(0 h)group.图6 免疫细胞化学检测缺氧对CHOP蛋白表达水平的影响
讨论
组织缺氧是指因氧气供应不足或用氧障碍,而导致组织代谢、功能和形态结构发生异常变化的病理过程[9-10]。心肌缺氧可导致以细胞坏死和凋亡为特征的心肌细胞损伤。近年研究表明,ERS相关的凋亡途径与缺氧诱发细胞凋亡密切相关[11-12]。UPR保护性机制主要由2条相对独立的内质网跨膜分子介导信号通路来调控:一条是IRE1和ATF6途径,即IRE1和ATF6通过诱导内质网分子伴侣使未折叠或错误折叠蛋白恢复正确构像,终止ERS和细胞凋亡;另一条是PERK途径,即PERK在翻译水平抑制蛋白合成,减少未折叠蛋白,最终促进ERS恢复,减少细胞凋亡。有实验表明ATF6和IRE1通路参与了缺氧诱发的心肌细胞ERS及凋亡。本实验在体外培养的乳鼠心肌细胞上发现,PERK通路在缺氧心肌细胞损伤中起到重要的作用,缺氧不仅能够在内质网应激早期激活UPR的PERK通路,对心肌细胞起到保护作用,当内质网应激时间延长,还能够激活其下游的凋亡信号分子,激活凋亡通路,引发心肌细胞凋亡。
本实验将原代培养的乳鼠心肌细胞置于密闭的恒温缺氧仓内,通入95%N2/5%CO2混合气体,氧气含量少于0.5%,模拟体内心肌细胞缺氧环境。以ATP含量作为细胞活力的检测指标,结果显示,随着缺氧时间的延长,细胞活力明显降低,但在缺氧早期,大约4 h,细胞活力有所逆转。多参数检测细胞凋亡的实验同样发现,在缺氧时间延长至12 h,无论从细胞的核亮度,还是线粒体膜电位及肌动蛋白含量的改变上来看,心肌细胞均表现出明显的凋亡特征,但在缺氧早期表现出轻微逆转。这说明在内质网应激早期,有一种保护性作用的存在对抗由缺氧引起的心肌细胞损伤。本实验设计不同缺氧时间体现了内质网应激对心肌细胞造成损伤的不同。内质网分子伴侣GRP78作为内质网稳态的感受器,在监测内质网中未折叠蛋白质的聚集和内质网应激的激活过程中发挥重要作用。钙网蛋白为ER中主要的钙结合伴侣蛋白,是ER贮存的重要应激蛋白和ERS的重要标志。本实验中,缺氧1 h,就能上调GRP78和钙网蛋白的表达,缺氧4、8 h与正常组表达量相当,缺氧12、24 h显著低于正常组,提示缺氧1 h后导致了内质网应激反应发生。有文献多次报道称,在缺氧模型中,GRP78和钙网蛋白作为内质网应激的诱导因子,在应激前期被激活表达[13-14]。上述实验现象与文献报道一致,表明本实验的缺氧模型是成功的。
eIF2α是ERS中PERK通路直接的调控因子,PERK通过催化eIF2α磷酸化,使GDP-eIF2α不能转化为GTP-eIF2α,不能形成GTP-eIF2α-MettRNA/40S复合体,结果Met-tRNA的翻译被终止,蛋白合成下降,未折叠蛋白减少等,最终促进ERS恢复,减少细胞凋亡。Boyce等[15]报道,p-eIF2α去磷酸化抑制剂salubrinal通过激活UPR具有对抗ERS相关的PC-12细胞凋亡的作用。本实验室前期工作也证实在原代培养的 Wistar大鼠心肌细胞上,salubrinal具有对抗ERS特异性诱导剂衣霉素(tunicamycin)引起的心肌细胞损伤的作用,并且证实salubrinal的保护作用与eIF2α的磷酸化水平相关[16]。因此,salubrinal对细胞的保护作用可以间接证明PERK通路参与了ERS相关的细胞凋亡。本实验在缺氧诱导的原代培养的乳鼠心肌细胞上,发现提前给予salubrinal组ATP含量较单纯缺氧组有所回升,即抑制p-eIF2α的去磷酸化,对缺氧心肌细胞损伤有保护作用。以上结果提示,缺氧激活了UPR中的PERK通路。
PERK位于内质网内,作为未折叠蛋白反应中的重要因子,在发生内质网应激时与游离的GRP78结合而使自身蛋白暴露,发生磷酸化与二聚化。活化的PERK蛋白进一步使内质网外侧的eIF2α发生磷酸化修饰,一方面通过eIF2α磷酸化抑制蛋白合成,另一方面激活下游的ATF4,ATF4合成后转位至细胞核内,作为转录因子能够上调内质网分子伴侣蛋白并参与氨基酸转运蛋白质的转录表达,同样有助于恢复内质网稳态[17-18]。本实验在原代培养的心肌细胞缺氧模型上检测缺氧是否激活了PERK通路,结果发现:在缺氧4 h时,PERK和eIF2α磷酸化水平与对照组相比,都显著升高。ATF4作为peIF2α的下游分子,在缺氧后4 h表达上调,明显高于对照组。GADD153/CHOP是内质网应激特异的凋亡蛋白,属C/EBP转录因子家族成员。正常情况下,CHOP表达十分低,在内质网应激反应时,CHOP作为PERK通路的下游分子,在缺氧12 h,表达明显增加。在哺乳动物中,调节蛋白合成是一个十分复杂的过程,我们不能够排除其它小分子蛋白来调节蛋白合成的可能。但是我们可以推测,在缺氧早期,缺氧激活UPR中的PERK通路,并且通过PERK的磷酸化使eIF2α磷酸化,并且诱导ATF4表达上调,起到部分抑制未折叠蛋白合成、保护细胞的作用。但在缺氧后期,CHOP作为PERK通路的下游分子,诱发细胞心肌凋亡,是导致心肌细胞缺氧损伤的一个重要原因。阻断PERK通路,减少CHOP的表达,对于减少细胞凋亡的发生将起到决定性作用,有关这方面的讨论有待进一步研究。
综上所述,本实验在原代培养的大鼠心肌细胞中发现,UPR反应中PERK通路蛋白及其下游的凋亡信号在缺氧过程中被激活,并在缺氧心肌细胞中依次表达。即ERS相关的PERK通路参与了缺氧诱发的心肌细胞内质网应激及凋亡。针对PERK通路与缺氧诱导心肌细胞损伤关系的研究,将有望为心肌缺血性疾病的防治提供新靶点。
[1]Wencker D,Chandra M,Nguyen K,et al.A mechanistic role for cardiac myocyte apoptosis in heart failure[J].J Clin Invest,2003,111(10):1497-1504.
[2]Szegezdi E,Duffy A,O'Mahoney ME,et al.ER stress contributes to ischemia-induced cardiomyocyte apoptosis[J].Biochem Biophys Res Commun,2006,349(4): 1406-1411.
[3]Faitova J,Krekac D,Hrstka R,et al.Endoplasmic reticulum stress and apoptosis[J].Cell Mol Biol Lett,2006,11(4):488-505.
[4]Davenport EL,Morgan GJ,Davies FE.Untangling the unfolded protein response[J].Cell Cycle,2008,7(7): 865-869.
[5]Negoro S,Kunisada K,Tone E,et al.Activation of JAK/ STAT pathway transduces cytoprotective signal in rat acute myocardial infarction[J].Cardiovasc Res,2000,47(4): 797-805.
[6]Ron D,Walter P.Signal integration in the endoplasmic reticulum unfolded protein response[J].Nat Rev Mol Cell Biol,2007,8(7):519-529.
[7]Doroudgar S,Thuerauf DJ,Marcinko MC,et al.Ischemia activates the ATF6 branch of the endoplasmic reticulum stress response[J].J Biol Chem,2009,284(43):29735-29745.
[8]Thuerauf DJ,Marcinko M,Gude N,et al.Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes[J].Circ Res,2006, 99(3):275-282.
[9]Gasparetto M,Gentry T,Sebti S,et al.Identification of compounds that enhance the anti-lymphoma activity of rituximab using flow cytometric high-content screening[J].J Immunol Methods,2004,292(1-2):59-71.
[10]Natarajan R,Salloum FN,Fisher BJ,et al.Prolyl hydroxylase inhibition attenuates post-ischemic cardiac injury via induction of endoplasmic reticulum stress genes[J].Vascul Pharmacol,2009,51(2-3):110-118.
[11]Osada N,Kosuge Y,Ishige K,et al.Characterization of neuronal and astroglial responses to ER stress in the hippocampal CA1 area in mice following transient forebrain ischemia[J].Neurochem Int,2010,57(1):1-7.
[12]杨 鹏,杨成明,曾春雨,等.心肌梗死后心力衰竭小鼠心肌组织内质网应激相关凋亡途径的研究[J].中国病理生理杂志,2010,26(6):1069-1074.
[13]Minamino T,Kitakaze M.ER stress in cardiovascular disease[J].J Mol Cell Cardiol,2010,48(6):1105-1110.
[14]Flores-Diaz M,Higuita JC,Florin I,et al.A cellular UDP-glucose deficiency causes overexpression of glucose/oxygen-regulated proteins independent of the endoplasmic reticulum stress elements[J].J Biol Chem,2004,279(21):21724-21731.
[15]Boyce M,Bryant KF,Jousse C,et al.A selective inhibitor of eIF2α dephosphorylation protects cells from ER stress[J].Science,2005,307(5711):935-939.
[16]胡国梁,何昆仑,范 利,等.Salubrinal保护心肌细胞内质网应激凋亡的实验研究[J].军医进修学院学报,2010,31(5):483-485.
[17]Terai K,Hiramoto Y,Masaki M,et al.AMP-activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress[J].Mol Cell Biol,2005,25(21):9554-9575.
[18]Cybulsky AV,Takano T,Papillon J,et al.Role of the endoplasmic reticulum unfolded protein response in glomerular epithelial cell injury[J].J Biol Chem,2005,280 (26):24396-24403.
PERK signal pathway is involved in hypoxia-induced endoplasmic reticulum stress and apoptosis in cultured cardiac myocytes
LIU Chun-lei1,2,LI Xin1,LI Rui-jun1,HE Yun-yun1,2,HE Kun-lun1,WANG Li- li3
(1Department of Cardiology,General Hospital of Chinese PLA,Beijing 100853,China;2Medical School of Nankai University,Tianjin 300071,China;3Institute of Poisons and Drugs,Academy of Military Medical Sciences,Beijing 100850,China.E-mail:hekunlun2002@yahoo.com;wangll63@126.com)
AIM:To investigate the role of endoplasmic reticulum(ER)stress in the process of hypoxia-induced neonatal rat myocardial injury through PERK signal pathway.METHODS:Neonatal rat cardiac myocytes were randomly divided into control group and hypoxia 1 h,4 h,8 h,12 h and 24 h groups.Cell viability was evaluated by determining the intracellular content of ATP.Apoptosis was measured by high-content analysis(HCA)cell imaging system.The protein levels of GRP78,calreticulin,p-PERK,p-eIF2α,ATF4 and CHOP were detected by Western blotting at different time points.The primary cultured neonatal rat cardiac myocytes were treated with an agonist of PERK pathway salubrinal and the cell apoptosis was observed under hypoxia.RESULTS:In the early phase,hypoxia induced an increase in the expression of calreticulin and GPR78.In the middle phase of hypoxia,the levels of p-PERK,p-eIF2α and ATF4 were increased.In the later phase of hypoxia,increased CHOP level was also observed.Salubrinal effectively protected the cardiac myocytes from hypoxic injury.CONCLUSION:Hypoxia activates ER stress in cardiac myocytes and also activates PERK signal pathway.PERK signaling protects cardiac myocytes from hypoxic damage in the early stage and triggers apoptosis of the cells in the later phase.
Cardiomyocytes;Endoplasmic reticulum stress;Hypoxia;Unfolded protein response;Apoptosis
R363.2
A
10.3969/j.issn.1000-4718.2012.08.010
1000-4718(2012)08-1392-07
2012-03-02
2012-06-25
国家科技部国际科技合作重大专项课题(No.2006DFB32210)
△通讯作者Tel:010-66939685;何昆仑E-mail:hekunlun2002@yahoo.com;王莉莉E-mail:wangll63@126.com
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31 09:58:22
解放军医学杂志(2021年12期)2022-01-18 03:53:24
现代临床医学(2021年1期)2021-01-26 00:55:52
解放军医学院学报(2020年12期)2020-03-29 05:11:32
心肺血管病杂志(2019年9期)2019-12-09 08:34:02
中成药(2018年9期)2018-10-09 07:18:32
安徽医科大学学报(2016年12期)2017-01-15 14:21:55
中西医结合心脑血管病杂志(2016年20期)2016-03-01 04:20:32
中国当代医药(2015年33期)2015-03-01 02:09:08
中华皮肤科杂志(2014年3期)2014-12-19 12:54:50
