
固相萃取柱高效液相色谱法测定复方甘草含片中吗啡的含量
2012-08-09 03:02熊水莲
实用临床医学 2012年10期
熊水莲,侯 莉
(1.江西制药有限责任公司质量检验部,南昌330052;2.中药固体制剂制造技术国家工程研究中心,南昌330006)
复方甘草含片是临床上常用的祛痰镇咳复方制剂,主要成分有甘草浸膏、阿片粉、樟脑和八角香油,其中阿片在复方甘草含片中的剂量远小于阿片镇痛、镇静的常用剂量。阿片所含的主要是生物碱吗啡。本文通过建立固相萃取柱高效液相色谱法(HPLC)测定复方甘草含片中吗啡含量的方法,旨在控制复方甘草含片中吗啡的含量。
1 仪器与试药
1.1 仪器
北京赛多利斯BT25S电子天平(十万分之一);日本岛津LC-10AvT紫外检测器;日本岛津LC-10AT VP泵;千谱工作色谱工作站。
1.2 试药
复方甘草含片(江西制药有限责任公司,批号:1201121、1201131、1111251);吗啡对照品(中国药品生物制品检定所,批号:171201-200521;含量:100%),乙腈为色谱纯,磷酸二氢钾为分析纯,庚烷磺酸钠为分析纯,醋酸为分析纯,氨试液(按药典方法配制)。
2 方法与结果
2.1 色谱条件系统适用性试验
色谱柱为Alltima C8 150mm×4.6mm,流动相为0.002 5mol·L-1庚烷磺酸钠水溶液-0.05mol·L-1磷酸二氢钾-乙腈(5∶5∶2),流速:1.0 mL·min-1,柱温:室温,检测波长220 nm,进样量20 uL。理论板数按吗啡峰计算不低于1 000,吗啡峰与相邻色谱峰的分离度应符合要求。
固相萃取柱系统适用性试验:用十八烷基硅烷键合硅胶为填充剂;以供试品溶液的制备中相同的处理条件和洗脱条件试验,精密量取浓度为每1mL中含吗啡对照品0.1 mg的5%醋酸溶液0.5 mL,置处理好的固相萃取柱上,同法洗脱,用5 mL量瓶收集洗脱液至刻度,摇匀,作为系统适用性试验溶液。精密量取系统适用性试验溶液和含量测定项下的对照品溶液各10 uL,分别注入液相色谱仪,记录色谱图,按下列公式计算,系统适用性试验溶液结果应在 0.97~1.03 之间[1]。
式中Ax为系统适用性试验溶液吗啡峰面积;Ar为对照品溶液吗啡峰面积;Cx为系统适用性试验溶液浓度;Cr为对照品溶液浓度。
2.2 溶液的制备
1)对照品溶液制备:取吗啡对照品约25 mg,精密称定,置100 mL量瓶中,加含2%甲醇、5%醋酸的溶液使溶解并稀释至刻度,摇匀,制成对照品贮备液,备用。精密量取2mL对照品贮备液置50mL,加含2%甲醇、5%醋酸的溶液使溶解并稀释至刻度,摇匀,即得。
2)供试品溶液制备:取复方甘草含片粉末10片称重,精密称定,置磨口锥形瓶中,精密加水90mL,超声处理5min,精密加稀盐酸10mL,摇匀,超声处理20 min使吗啡溶解,取出,放冷,滤过;精密量取续滤液1mL,置固相萃取柱[取固相萃取柱1支,依次用甲醇-水(3∶1)15 mL 与水 5mL 冲洗,再用氨水溶液(pH值为9)冲洗至流出液pH值为9,滴加氨试液适量,使萃取柱内溶液的pH值为9(上柱前另取同体积的续滤液预先调试,以确定滴加氨试液的量),摇匀,待溶剂滴尽后,用水约20mL冲洗,用含2%甲醇、5%醋酸的溶液洗脱,用5 mL量瓶收集洗脱液至刻度,摇匀,即得。
2.3 测定方法
精密量取对照品溶液、供试品溶液各20 uL,照“2.1”项下色谱条件分别注入液相色谱仪,记录色谱图,按外标法计算,即得。
2.4 线性关系的考察
取对照品贮备液,置于10 mL量瓶中,用流动相稀释,制得浓度分别为 5﹑10 ﹑15 ﹑20﹑30 μg·mL-1;进样20 uL测定,以对照品浓度对峰面积积分值进行线性回归。结果得回归方程:A=1.42×105C-0.459 5,r=1,显示吗啡在5~30μg·mL-1范围内线性关系良好。
2.5 精密度试验
取复方甘草含片的供试品溶液 20μL,按上述色谱条件连续进样5次。测得主峰面积分别为993784、994359、995021、993951、994209,平均值为994265,计算RSD为0.1%。
2.6 重复性试验
取同一批号的复方甘草含片按“2.2”项下方法制备供试品溶液,按“2.1”项下所述色谱条件分别进样重复测定3次,计算平均含量为 0.39 mg·片-1,RSD为0.9%,说明重复性良好,
2.7 稳定性考察
取供试品溶液 1 份,分别于 0﹑2﹑4﹑6﹑8 h 分别进样20μL测定,吗啡峰面积基本不变,RSD为0.9%。
2.8 回收率实验
精密称量复方甘草含片粉末约0.54 g共5份,每份分别准确加入对照品溶液适量,按“2.2”项下方法制备供试品溶液,按“2.1”项所述色谱条件重复进样3次,计算加样回收率,结果见表1。
2.9 样品含量测定
精密称量各批号复方甘草含片约5.44 g,按“2.2”项下方法制备供试品溶液,按“2.1”项所述色谱条件分别进样测定,计算其含量,结果见表2。结果表明,该3批复方甘草含片中吗啡的含量均符合规定(0.36~0.44mg·片-1[2])。
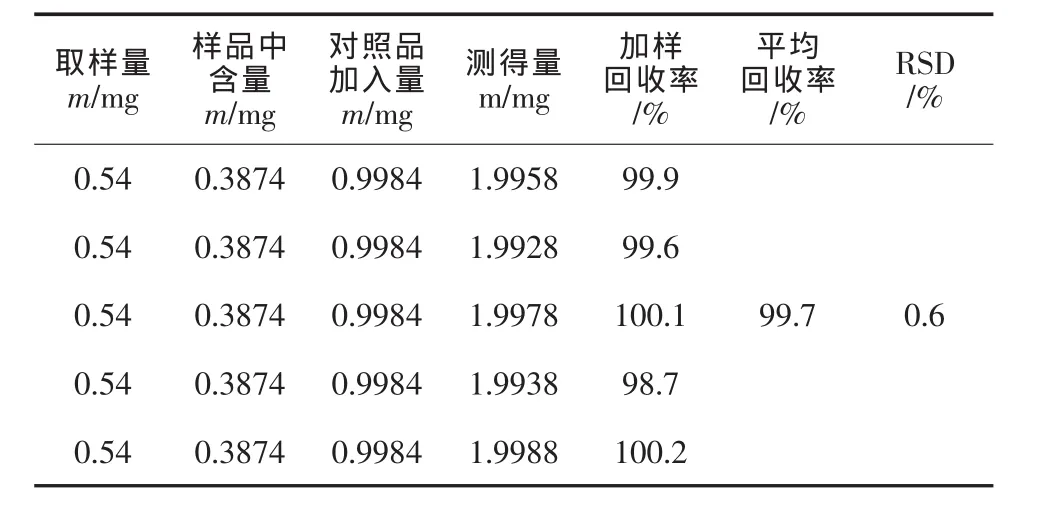
表1 回收率测定结果

表2 样品含量测定结果
3 讨论
3.1 流动相的选择
本方法中选用庚烷磺酸钠试剂,由于其是离子对试剂,离子对试剂用于高压液相萃取液相色谱中能改善峰形,分析结果重现性好;选用乙腈作为有机相是由于乙腈对柱子的损耗小,洗脱能力比甲醇强,而且不易出现鬼峰。
3.2 波长的选择
由于甲醇的截止峰的波长是190 nm,而乙腈的截止波长是210 nm,所以选择波长为220 nm。
3.3 进样量的选择
本实验采用的仪器是手动进样器进样,由于此仪器的最低定量环进样量为20μL,若使用微量进样器进10μL,易产生气泡且人为因素易产生误差,而采用20μL定量环进样,具有量样精密、快速、便捷等特点。所以本文采用20μL定样环进样。
3.4 色谱柱的选择
C18出峰时间慢,能达到很好的效果;C8出峰时间快,能节约时间,在本实验中测得是吗啡的含量,只要能达到分离的效果即可。故选用C8作为色谱柱。
用固相萃取方法处理复方甘草含片,简便、快速,样品处理时间短。由于样品中吗啡的含量极少,若分离提取和洗涤次数较多,吗啡含量会损失,吗啡在碱性中也容易流失,中国药典[2]中检测的方法流程过于复杂、繁锁,提取的结果误差比较大,而采用本法测定含量更准确可靠,操作起来简捷,可用于复方甘草含片生产的质量控制。
[1]曾勇固.固相萃取技术-HPLC测定复方甘草片中吗啡的含量[J].色谱,1998,16(3):291.
[2] 中华人民共和国国家药典委员会.中国药典 (二部)[M].北京:中国医药科技出版社,2010:574.
猜你喜欢
家庭影院技术(2021年2期)2021-03-29
食品工程(2020年4期)2021-01-20
中国油脂(2020年3期)2020-04-10
建材发展导向(2019年10期)2019-08-24
淄博师专论丛(2019年1期)2019-04-04
益寿宝典(2018年16期)2018-01-27
无机化学学报(2016年8期)2016-12-06
铁道通信信号(2016年1期)2016-06-01
化学分析计量(2016年1期)2016-03-14
中国药业(2014年17期)2014-05-26
