
不同锰氧化度水钠锰矿的XPS研究
2012-07-31 13:04:10赵巍刘凡冯雄汉谭文峰
中南大学学报(自然科学版) 2012年2期
赵巍 ,刘凡,冯雄汉,谭文峰,
(1. 西北农林科技大学 黄土高原土壤侵蚀与旱地农业国家重点实验室,陕西 杨凌,712100;2. 中国科学院 水利部水土保持研究所黄土高原土壤侵蚀与旱地农业国家重点实验室,陕西 杨凌,712100;3. 华中农业大学 农业部亚热带农业资源与环境重点开放实验室,湖北 武汉,430070)
环境中的重金属铅对动植物具有毒性,特别是对人的神经系统有损害作用。大量研究表明,吸附是一种能使铅从液相转变为固相的重要过程,可影响铅的分布、迁移和生物有效性[1]。氧化锰矿物广泛分布于土壤、沉积物和海洋锰结核中,其电荷零点(PZC)低,比表面积大,负电荷量高,表面活性强,是土壤与沉积物中吸附铅的重要载体[2-4]。研究铅与氧化锰的作用机理,对于深入理解铅在环境中形态与转化、固定与释放的地球化学行为具有重要意义[5]。层状水钠锰矿是表生环境中最常见的锰氧化物,并且许多氧化锰矿物可以为母体直接或间接转变而成[6-7]。KMnO4在回流条件下被浓盐酸还原而得到的水钠锰矿通常称为酸性水钠锰矿[8-9],其MnO6八面体层主要是由Mn(IV)O6和八面体空位共同构成,有时含有部分Mn(III)O6,且在空位的上方或下方有部分 Mn2+或 Mn3+存在[10-11]。水钠锰矿结构中的锰氧八面体空位为负电荷的产生,Pb,Zn,Cu,Cd和Ni等重金属离子吸附,Co2+和Cr3+等氧化及矿物转化等方面起着十分重要的作用[12-18]。扩展X线吸收精细结构光谱(EXAFS)和X线衍射图谱(XRD)分析表明:Pb2+以八面体配位离子构型吸附于水钠锰矿表面,其中大部分Pb2+与八面体空位通过共用氧原子而在表面形成三齿共角配位[19-20]。但目前人们对水钠锰矿结构中八面体空位的具体分布状况并不清楚。X线光电子能谱是一种重要的表面分析技术,它不仅能探测样品表面的化学组成,而且可以确定各元素的化学状态,不损坏样品,光电子逃逸深度为0.5~10 nm,因此,其在化学、材料科学及表面科学中应用广泛。通过对Cr3+,H3AsO3以及腐殖酸盐与水钠锰矿反应过程中的中间产物进行XPS测试,并利用图谱分峰拟合的方法得到中间产物表层Mn和O的不同化学状态,可研究其反应动力学机理[21-23]。在此,本文作者通过X线光电子能谱技术对不同锰氧化度水钠锰矿表层Mn和O的化学状态进行分析,以便为水钠锰矿八面体空位的分布状况以及表面化学性质研究提供进一步结构信息。
1 实验
1.1 试剂与仪器
实验所用试剂均为分析纯 (AR)试剂,购于国药集团化学试剂有限公司。试剂的配置和实验用水均为去离子水,电导率小于2.0 μS/cm。
仪器为D/Max-3B X线衍射仪、XSAM800多功能电子能谱仪、Quantachrome Autosorb-1型全自动比表面和孔径分布分析仪。
1.2 水钠锰矿的合成
用300~400 mL去离子水溶解0.2 mol KMnO4于三角瓶中,将其在恒温油浴加热下煮沸(110 ℃即可),开启强力搅拌后,按0.7 mL/min的速率逐滴分别加入45.0,53.3,53.3,45.0和66.7 mL 6 mol/L及35.0 mL 12 mol/L HCl溶液,滴加完毕后继续反应30 min,产物在60 ℃时老化处理12 h[24],依次得到样品HB1,HB2,HB3,HB4,HB5 和 HB6。
1.3 样品表征
1.3.1 X线衍射分析(XRD)
将合成的水钠锰矿按粉末压片法进行 X线衍射(XRD)分析。测试条件为:Fe Kα辐射(波长λ=0.193 73 nm),管压为40 kV,管流为20 mA,扫描速度为0.02(°)/(0.4 s)。
1.3.2 X线光电子能谱分析(XPS)
矿物样品的元素电子结合能在XSAM800多功能电子能谱仪上用Mg KαX线分析,真空压为2×10-7Pa。 Mn和O元素的高精度窄区谱用XPSpeak41 软件进行多峰高斯拟合分离重叠峰。采用污染碳 C1s(EBE=284.62 eV)作为荷电校正标准。
1.3.3 比表面积分析(SSA)
样品的比表面在Quantachrome Autosorb-1型全自动比表面和孔径分布分析仪上测定。样品测试前,在80 ℃进行脱气处理约8 h,以除去水和其他吸附质。
1.3.4 水钠锰矿的化学组成分析
称取0.150 0 g样品,加入50 mL盐酸羟胺 (0.25 mol/L)溶解,定容至1 L;然后,用火焰分光光度计测定其K+含量,用原子吸收分光光度计测定其Mn离子含量。样品结构含水量依据电荷平衡和质量平衡计算得到。锰的氧化度采用草酸法测定[25]。做3个平行实验,结果取平均值。
2 结果与分析
图1所示是合成的水钠锰矿样品的X线衍射图谱。由图1可以看出:它们均为单相水钠锰矿,仅微结构存在差异[26],样品的特征衍射峰波长分别为 0.721,0.361,0.246和0.142 nm,这与JCPDS卡No.23-1239的酸性条件下合成的水钠锰矿的特征衍射峰波长基本一致。
合成水钠锰矿的化学组成、锰氧化度和比表面积见表1。从表1可见:随着锰氧化度的增大,样品的比表面积呈整体减少的趋势,并且呈显著负相关(相关系数r=-0.882 0,自由度n=6,显著水平α0.05=0.811 4)。图2所示为供试样品的Mn 2p和O 1s光电子能谱窄区谱,Mn 2p3/2谱峰位置出现在 642.10~642.60 eV范围内,基本在锰氧化物标准 Mn 2p3/2谱峰峰位(640.9~642.5 eV)范围内;O 1s谱峰在 530.17~530.77 eV之间,在金属氧化物标准 O 1s谱峰峰位(528.05~531.05 eV)范围内。
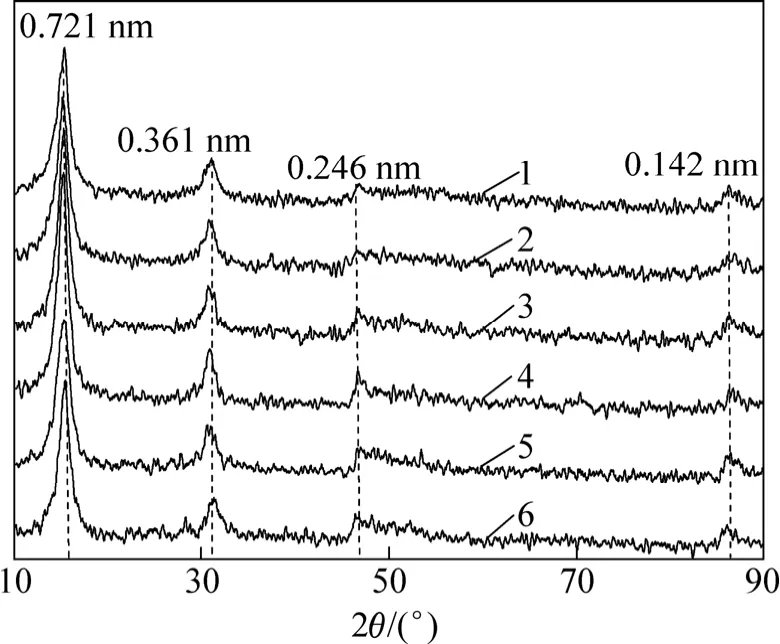
图1 供试样品的X线衍射图Fig.1 XRD patterns of samples
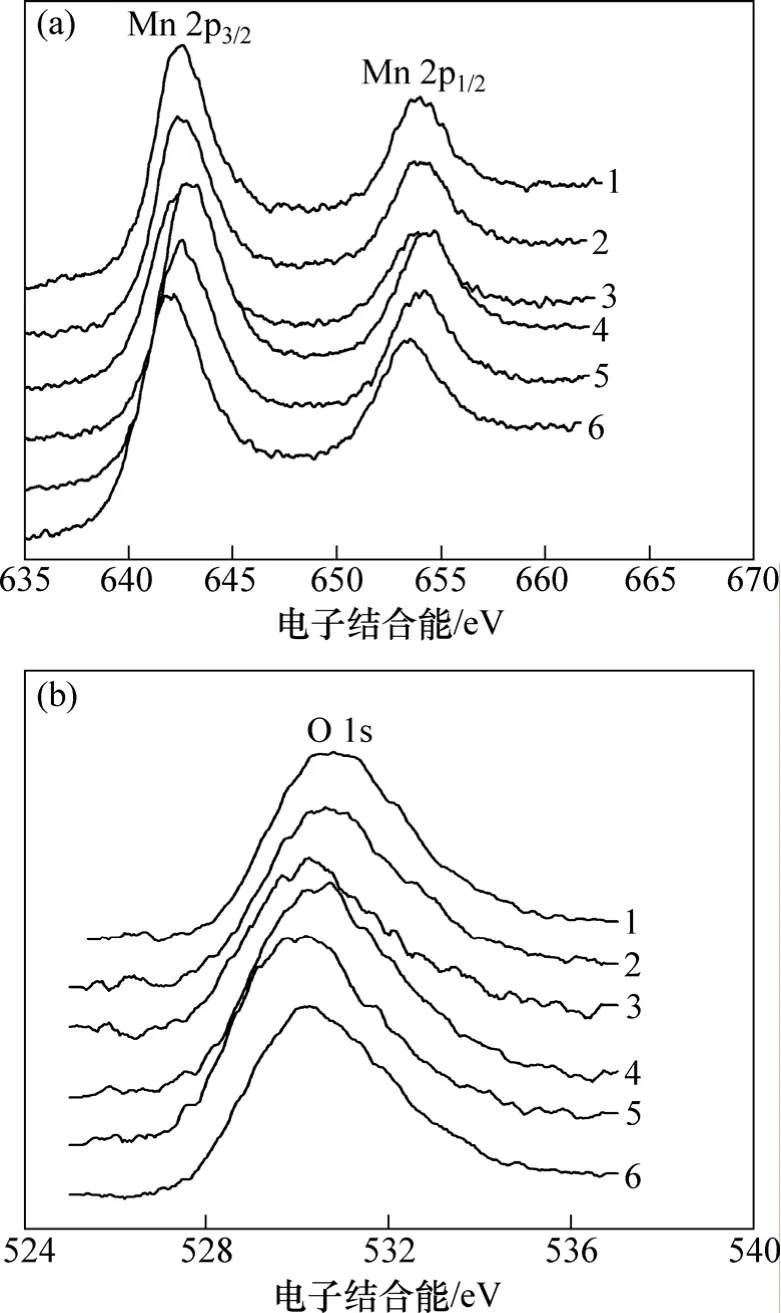
图2 供试样品的Mn 2p和O 1s光电子能谱Fig.2 XPS spectra of Mn 2p and O1s regions for surface of samples
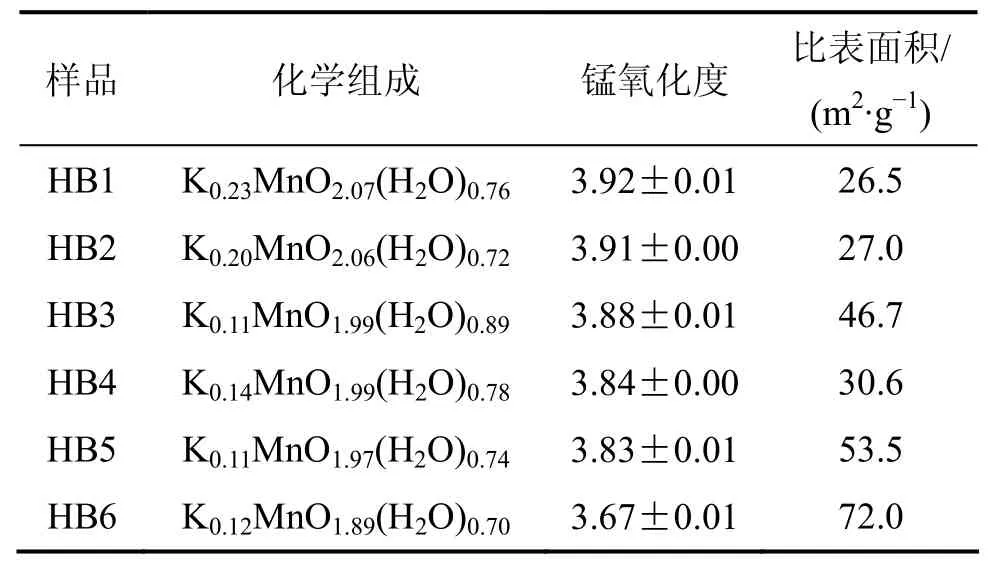
表1 供试样品的化学组成、锰氧化度和比表面积Table 1 Chemical composition, AOS and SSA of samples
3 讨论
供试样品层内只含有 Mn3+和 Mn4+,并且样品HB1,HB2,HB3,HB4和HB5的层间Mn2+或Mn3+含量较少,而样品 HB6层间可能含有部分 Mn2+或Mn3+。此外,锰氧化度高的样品其结构八面体空位较多,结合的—OH相应较多[26]。
3.1 Mn 2p3/2窄区谱图
从图2(a)可以看出:Mn 2p3/2谱峰不对称,Mn应该存在1种以上的化学状态。以样品HB1为例,若以的激发终态对Mn 2p3/2窄区谱进行分峰拟合,得到的Mn3+和Mn4+的摩尔数分数分别为47.06%和52.94%,其中,分峰拟合过程中采用的 Mn3+和 Mn4+激发终态所对应的电子结合能见文献[21]。但实际上,该样品锰氧化度为 3.92,设 Mn3+和 Mn4+占总锰的摩尔数分数分别为m和n,则m+n=1。由于锰平均氧化度为锰氧化物中锰的平均价态,故 3m+4n=3.92,可算出样品 HB1中Mn3+占总锰的摩尔数分数为8%,因此,以Mn的不同价态对Mn 2p3/2窄区谱进行拟合得到的Mn3+的摩尔数分数(47.06%)与实际不符。由于在光电子能谱测试条件下,真空度高,造成样品结构表面部分Mn离子的配位数减少。这些不饱和配位Mn离子是一种特殊的存在状态,因此,本文以饱和配位和不饱和配位这2种Mn的化学状态对Mn 2p3/2窄区谱进行分峰拟合,由此得到的样品中Mn的2种化学状态的电子结合能分别为641.72~642.70 eV和643.84~644.99 eV (表2),其摩尔数分数分别为 83.79%~91.69%和8.31%~17.21%。对于不饱和配位的Mn离子,由于其配位数减少,在Mn3+周围的电子云密度将减小,其外层电子对内层电子的屏蔽作用减弱,核对内层电子的束缚能力加强,从而导致内层电子的结合能增大[27]。因此,结构表面不饱和配位的Mn离子对应高结合能谱峰,而饱和配位的Mn离子对应的是低电子结合能谱峰。当样品的锰氧化度由3.92减小到3.67时,其对应的表面不饱和配位的Mn离子摩尔数分数由8.31%增加到17.21%。这可能是由于锰氧化度高的样品,其—OH含量较高,晶粒间相互吸附较强,较易发生团聚。这类似于纳米晶粒通过其表面的氢键作用相互吸附而发生团聚[28]。因此,虽然供试样品的晶粒平均相差不大[26],但由于团聚作用,导致其比表面积随锰氧化度的增大而减小,并且供试样品锰氧化度与其比表面积存在显著负相关。当样品的锰氧化度增大时,其比表面积减少,结构中暴露的表面不饱和配位Mn离子减少,因此,所测得的表面不饱和配位Mn离子含量相应减少,并且样品表面不饱和配位Mn离子含量与其比表面积呈显著正相关 (r=0.873 7,n=6,α0.05=0.811 4)。

表2 样品表面结构中不同化学状态Mn的摩尔数分数Table 2 Contents of Mn with different chemistry states on surface of samples
3.2 O1s窄区谱图
O元素的XPS谱峰也不对称 (图2(b)),表明晶粒表面氧的化学状态也并不为单一的晶格氧,还应含有羟基氧和水分子中氧等[28],因此,采用晶格氧、羟基氧和水分子中的氧这3种化学状态对样品表面氧物种的O1s窄区谱进行分峰拟合,其结果见表3。由于H的电负性(χH=2.1)较 Mn 的(χMn=1.55) 大,与 H 结合的氧离子周围的电子云密度比与Mn结合的氧离子周围的电子云密度小,其外层电子对内层电子的屏蔽作用较弱,核对内层电子的束缚能力较强,因此,与H结合的氧的O1s电子结合能较高,从而晶格氧、羟基氧和水分子中氧的O1s电子结合能依次增大。供试样品中的O1s电子结合能为529.66~530.13,531.41~531.84和532.97~ 533.75 eV,分别对应的是其结构中晶格氧、羟基氧和水分子中的氧3种化学状态,它们的摩尔数分数分别为 50.44%~65.05%,24.90%~39.27% 和8.07%~ 12.63%,对于锰氧化度较小的样品,由于其结构中空位数量较少,相应的位于空位上的羟基也较少,从而其结构中羟基氧摩尔数分数较小。
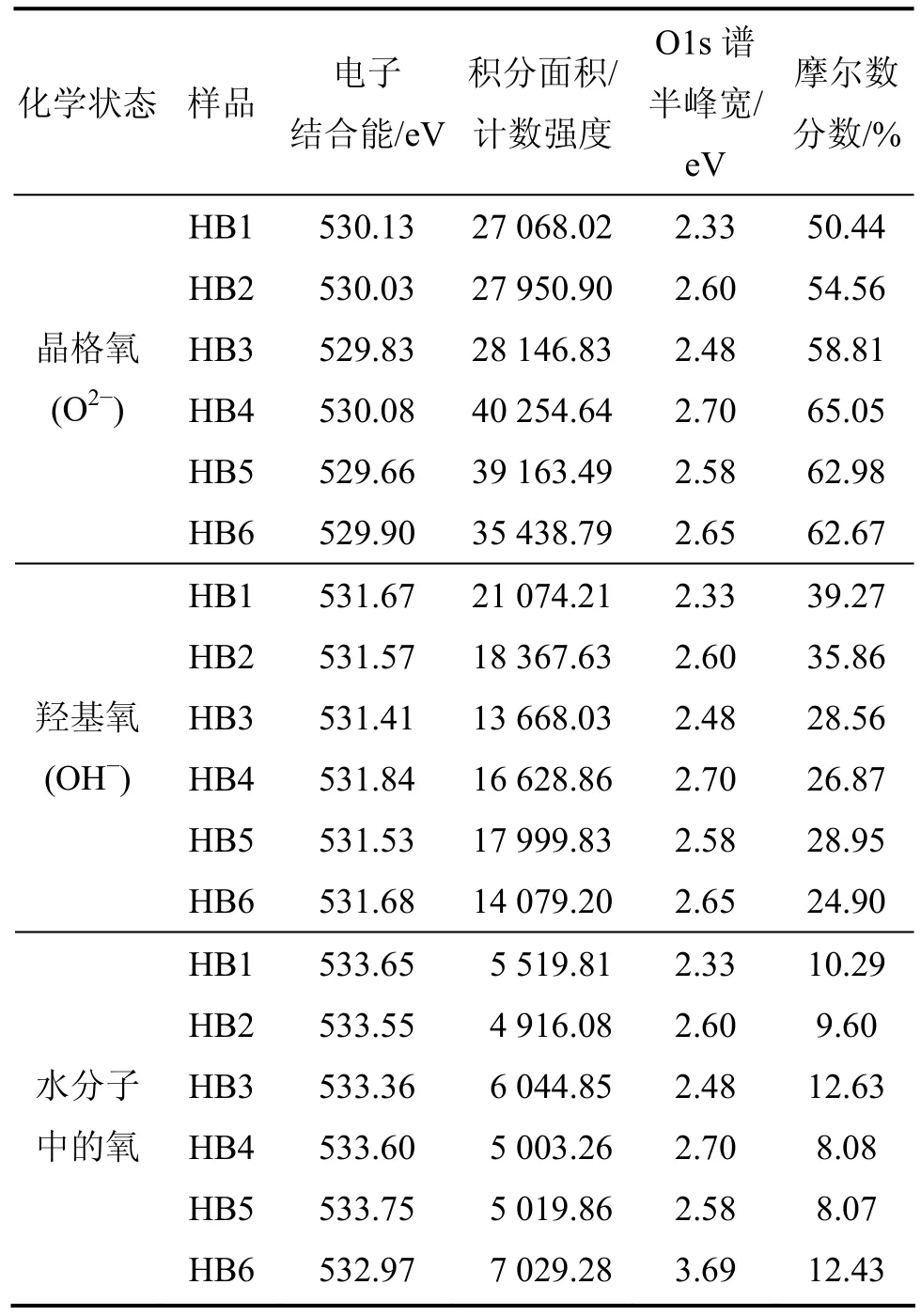
表3 供试样品表面结构中氧不同化学状态的摩尔数分数Table 3 Molar fraction of oxygen atoms with different chemistry states on surface of samples
3.3 水钠锰矿空位分布的结构模型
据文献[8],样品HB1(锰氧化度为3.92)对Pb2+的最大吸附量为2.457 mol/kg[26]。Manceau等[16]认为Pb2+主要吸附于水钠锰矿结构中八面体空位的上、下方,并且当空位含量最大时,仅为总锰含量的 16.7%,水钠锰矿的摩尔质量为100 g/mol。假设Pb2+仅能吸附于八面体空位的上方或下方,则水钠锰矿对Pb2+的最大吸附量约为1.670 mol/kg,而样品HB1对Pb2+的最大吸附量达到2.457 mol/kg,约为其1.5倍。由此可见:Pb2+应该可同时吸附于八面体空位的上方和下方。因此,Pb2+的吸附容量约为空位含量的2倍,由此可估算出样品 HB1结构中空位含量与总锰含量的表观比值为0.122 9(假设水钠锰矿的摩尔质量为100 g/mol)。另外,考虑到样品结构层间几乎不存在Mn3+,且层内Mn3+较易发生歧化反应而产生空位[16],于是,结合上述实验结果,构建1个结构模型A (图3(a))。图中黑色粗虚线区域所包含的MnO6单元为周期重复单元,其锰氧化度为3.923,与实验结果3.920相吻合;空位含量与总锰含量的比值为 0.153 8,也与实验结果0.122 9较接近。同样,根据文献[26]得到样品HB2,HB3,HB4,HB5和HB6的锰氧化度和对Pb2+的吸附容量,也可以分别构建出结构模型B,C,D和E(皆忽略层间Mn3+)(见图3(b)~(e))。由模型A至E,水钠锰矿结构中沿 (110)方向上的含 Mn3+的 MnO6八面体链中Mn3+含量增大,结构中空位含量相应减少。由于周期重复单元上、下方化学环境相同,所以,只以其1个周期重复单元进行讨论。
对于结构中的氧,可分为与空位相邻 (羟基氧)和不与空位相邻(晶格氧)2种化学状态。供试样品及所有结构模型的锰氧化度,空位含量与总锰含量的比值及不同化学状态O的含量如表4所示。对于供试样品,其锰氧化度为3.670~3.920,空位含量与总锰含量的比值为0.066 0~0.122 9,结构中羟基氧和晶格氧摩尔数分数分别为29.23%~43.77%和56.23%~70.77%;对于构建的结构模型,其锰氧化度为3.786~3.925,空位含量与总锰含量的比值为0.071 43~0.153 80,结构中羟基氧和晶格氧摩尔数分数分别为 20%~40%和60%~80%(由周期重复单元中的不同化学状态的氧原子数计算而得)。由表4可看出:样品HB1,HB2和HB3分别与结构模型A,B和C的锰氧化度以及结构中羟基氧和晶格氧摩尔数分数非常接近,只是样品中空位含量与总锰含量的比值皆比结构模型的低,这可能是吸附后样品中并不是所有的空位都被Pb2+占据或有部分Pb2+吸附于层结构边面所致,因此,样品HB1,HB2和HB3的层结构可能分别对应于结构模型A,B和C。样品HB4和HB5除了其空位含量与总锰含量的比值较结构模型C和D的偏低外,它们的锰氧化度(3.84和3.83)、结构中羟基氧 (29.23%和31.49%)和晶格氧 (70.77%和 68.51%)的摩尔数分数皆介于结构模型C和D的之间 (结构模型C和D的锰氧化度分别为3.875和3.818,羟基氧摩尔数分数分别为33.30%和 25.00%,晶格氧摩尔数分数分别为 66.70%和75.00%),因而,样品HB4和HB5的结构可能对应于结构模型C和D的混合态。至于样品HB6,除了其空位含量与总锰含量的比值与结构模型E的相比略偏低外,锰氧化度、羟基氧和晶格氧的摩尔数分数皆远比结构模型E的低,这有可能是样品HB6结构中含有大量的层间Mn3+所导致。
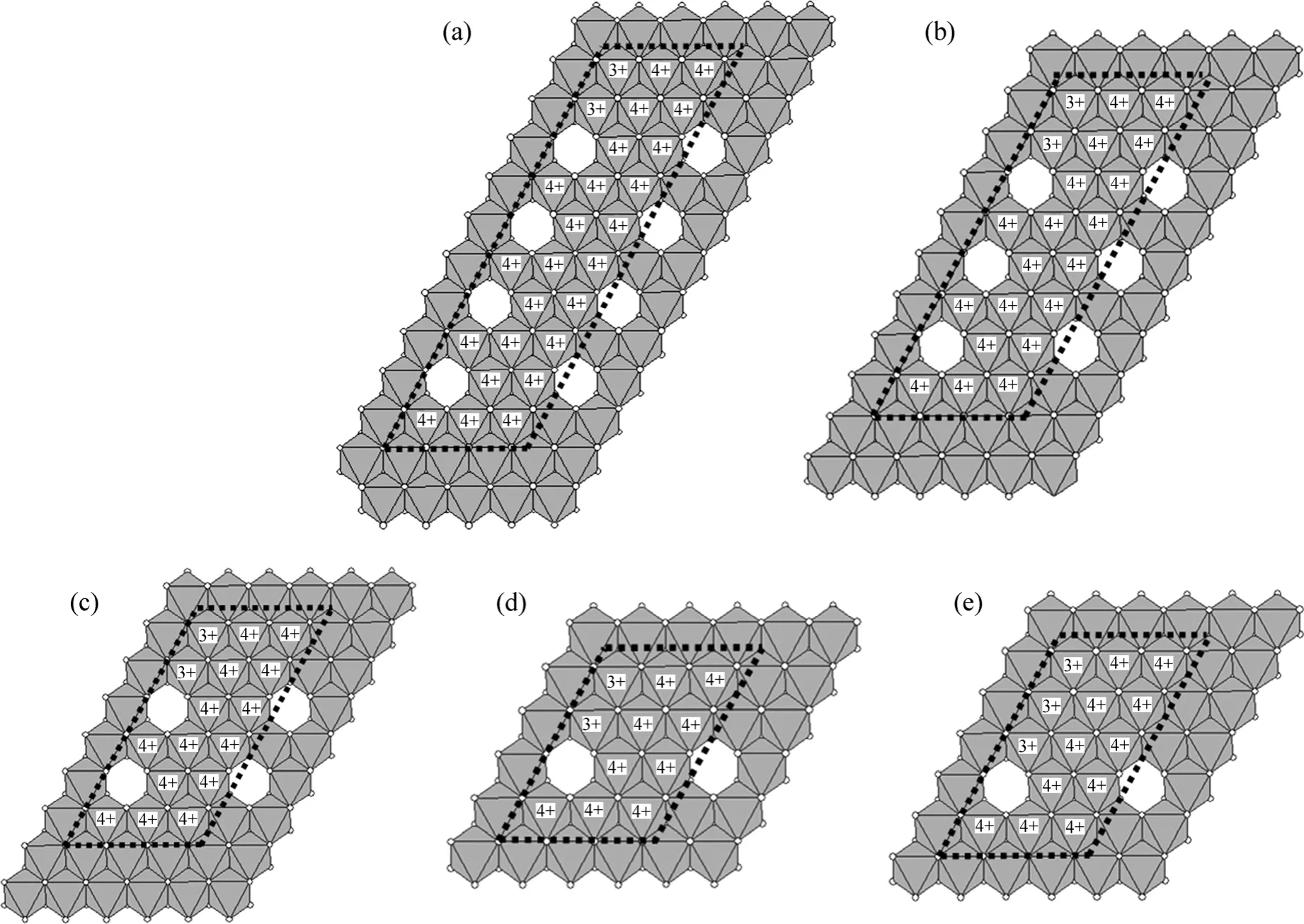
图3 水钠锰矿结构模型Fig.3 Structure models for birnessites A, B, C, D and E
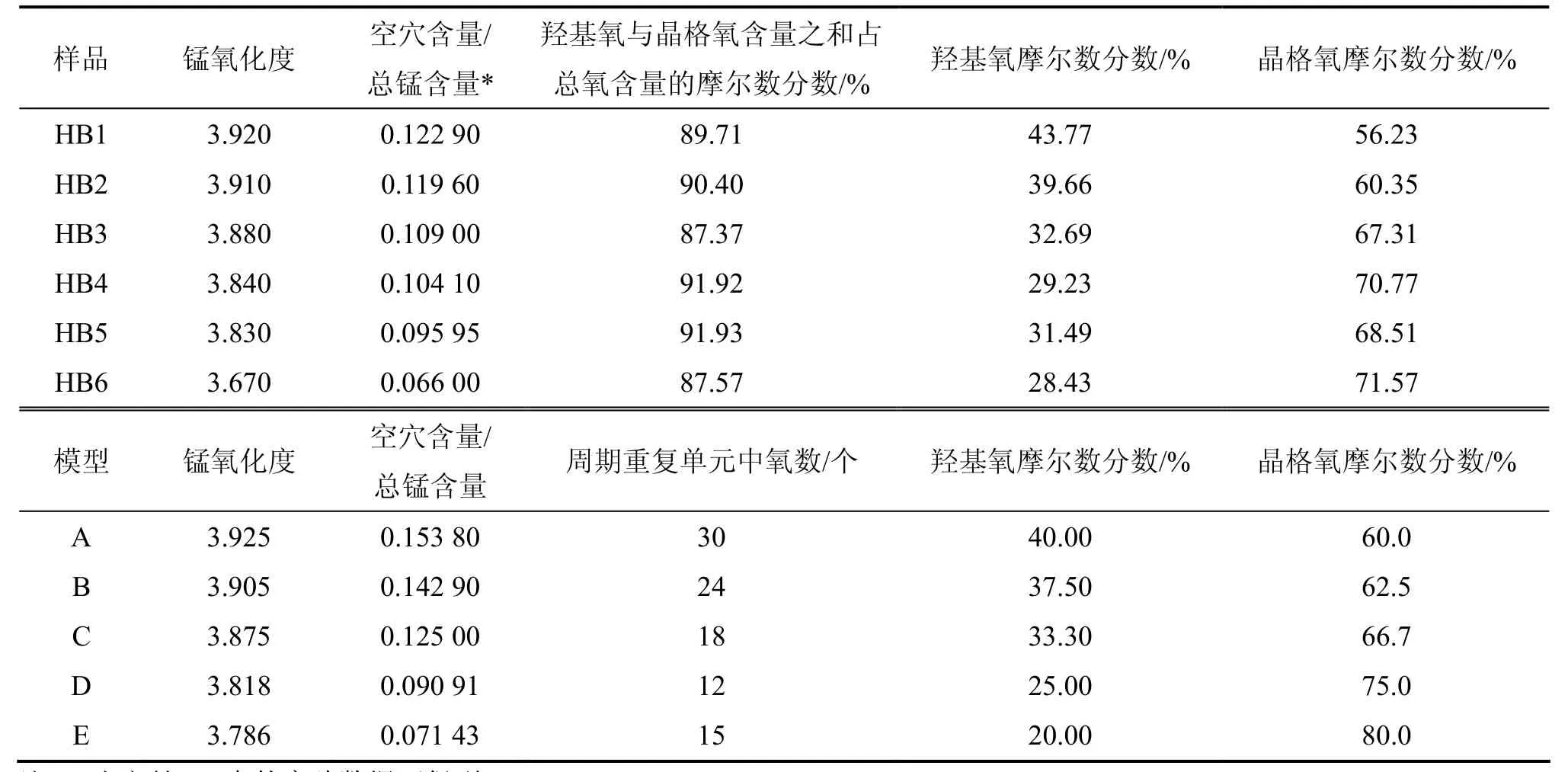
表4 供试水钠锰矿与结构模型的结构参数比较Table 4 Parameters for tested birnessites and structure models
4 结论
(1) 在高真空测试环境下,不同锰氧化度水钠锰矿结构中,Mn离子存在饱和配位和不饱和配位2种化学状态,其摩尔数分数分别为 83.79%~91.69%和8.31%~17.21%,锰氧化度高的水钠锰矿,八面体空位上结合的—OH较多,由于晶粒间的团聚作用而导致表面不饱和配位Mn的含量减少。
(2) 在不同锰氧化度水钠锰矿结构中,O存在晶格氧、羟基氧和水分子中的氧3种化学状态,其摩尔数分数分别为 50.44%~65.05%,24.90%~39.27%和8.07%~12.63%。锰氧化度较高的水钠锰矿,其八面体空位上结合的—OH较多,从而结构中羟基氧的含量增加。
(3) 水钠锰矿沿(110)方向上含Mn3+的MnO6八面体链中Mn3+含量增大,结构中空位含量相应减少。
[1]Xu Y, Boonfueng T, Axe L, et al. Surface complexation of Pb(II)on amorphous iron oxide and manganese oxide: Spectroscopic and time studies[J]. Journal of Colloid and Interface Science,2006, 299(1): 28-40.
[2]Mckenzie R M. The adsorption of lead and other heavy metals on oxides of manganese and iron[J]. Australian Journal of Soil Research, 1980, 18(1): 61-73.
[3]Post J E. Manganese oxide minerals: Crystal structures and economic and environmental significance[J]. Proceedings of the National Academy of Sciences of the United States of America,1999, 96(7): 3447-3454.
[4]O’reilly S E, Hochella M F. Lead sorption efficiencies of natural and synthetic Mn and Fe-oxides[J]. Geochimica et Cosmochimica Acta, 2003, 67(23): 4471-4487.
[5]Matocha C J, Elzinga E J, Sparks D L. Reactivity of Pb(II) at the Mn(III, IV) (oxyhydr)oxide-water interface[J]. Environmental Science & Technology, 2001, 35(14): 2967-2972.
[6]Golden D C, Chen C C, Dixon J B. Transformation of birnessite to buserite, todorokite, and manganite under mild hydrothermal treatment[J]. Clays and Clay Minerals, 1987, 35(4): 271-280.
[7]Tu S, Racz G J, Goh T B. Transformations of synthetic birnessite as affected by pH and manganese concentration[J]. Clays and Clay Minerals, 1994, 42(3): 321-330.
[8]Mckenzie R M. The synthesis of birnessite, cryptomelane,and some other oxides and hydroxides of manganese[J].Mineralogical Magazine, 1971, 38(296): 493-502.
[9]Villalobos M, Toner B, Bargar J, et al. Characterization of the manganese oxide produced byPseudomonas putidastrain MnB1[J]. Geochimica et Cosmochimica Acta, 2003, 67(14):2649-2662.
[10]Webb S M, Tebo B M, Bargar J R. Structural characterization of biogenic Mn oxides produced in seawater by the marinebacillus sp.strain SG-1[J]. American Mineralogist, 2005, 90(8/9):1342-1357.
[11]Villalobos M, Lanson B, Manceau A, et al. Structural model for the biogenic Mn oxide produced byPseudomonas putida[J].American Mineralogist, 2006, 91(4): 489-502.
[12]Burns R G. The uptake of cobalt into ferromanganese nodules,soils, and synthetic manganese (IV) oxides[J]. Geochimica et Cosmochimica Acta, 1976, 40(1): 95-102.
[13]Manceau A, Charlet L. X-ray absorption spectroscopic study of the sorption of Cr(III) at the oxide-water interface : I. Molecular mechanism of Cr(III) oxidation on Mn oxides[J]. Journal of Colloid and Interface Science, 1992, 148(2): 425-442.
[14]Appelo C A J, Postma D. A consistent model for surface complexation on birnessite (-MnO2) and its application to a column experiment[J]. Geochimica et Cosmochimica Acta, 1999,63(19/20): 3039-3048.
[15]Lanson B, Drits V A, Feng Q, et al. Structure of synthetic Na-birnessite: Evidence for a triclinic one-layer unit cell[J].American Mineralogist, 2002, 87(11/12): 1662-1671.
[16]Manceau A, Lanson B, Drits V A. Structure of heavy metal sorbed birnessite. Part III: Results from powder and polarized extended X-ray absorption fine structure spectroscopy[J].Geochimica et Cosmochimica Acta, 2002, 66(15): 2639-2663.
[17]Toner B, Manceau A, Webb S M, et al. Zinc sorption to biogenic hexagonal-birnessite particles within a hydrated bacterial biofilm[J]. Geochimica et Cosmochimica Acta, 2006, 70(1):27-43.
[18]Peacock C L, Sherman D M. Sorption of Ni by birnessite:Equilibrium controls on Ni in seawater[J]. Chemical Geology,2007, 238(1/2): 94-106.
[19]Lanson B, Drits V A, Gaillot A C, et al. Structure of heavy-metal sorbed birnessite: Part 1. Results from X-ray diffraction[J].American Mineralogist, 2002, 87(11/12): 1631-1645.
[20]Villalobos M, Bargar J, Sposito G. Mechanisms of Pb(II)sorption on a biogenic manganese oxide[J]. Environmental Science & Technology, 2005, 39(2): 569-576.
[21]Nesbitt H W, Canning G W, Bancroft G M. XPS study of reductive dissolution of 7Å birnessite by H3AsO3, with constraints on reaction mechanism[J]. Geochimica et Cosmochimica Acta, 1998, 62(12): 2097-2110.
[22]Banerjee D, Nesbitt H W. Oxidation of aqueous Cr(III) at birnessite surfaces: constraints on reaction mechanism[J].Geochimica et Cosmochimica Acta, 1999, 63(11/12):1671-1687.
[23]Banerjee D, Nesbitt H W. XPS study of dissolution of birnessite by humate with constraints on reaction mechanism[J].Geochimica et Cosmochimica Acta, 2001, 65(11): 1703-1714.
[24]Kijima N, Yasuda H, Sato T, et al. Preparation and characterization of open tunnel oxide [alpha]-MnO2precipitated by ozone oxidation[J]. Journal of Solid State Chemistry, 2001,159(1): 94-102.
[25]Zhao W, Cui H J, Feng X H, et al. Relationship between Pb2+adsorption and average Mn oxidation state in synthetic birnessites[J]. Clays and Clay Minerals, 2009, 57(3): 338-345.
[26]Grosvenor A P, Kobe B A, Biesinger M C, et al. Investigation of multiplet splitting of Fe 2p XPS spectra and bonding in iron compounds[J]. Surface and Interface Analysis, 2004, 36(12):1564-1574.
[27]冯拉俊, 刘毅辉, 雷阿利. 纳米颗粒团聚的控制[J]. 微纳电子技术, 2003, 40(7): 536-540.FENG La-jun, LIU Yi-hui, LEI A-li. The controlling of nanoparticle agglomerates[J]. Micronanoelectronic Technology,2003, 40(7): 536-540.
[28]姜洪泉, 王鹏, 钱恒泽. 低量 Yb3+掺杂的 TiO2复合纳米粉体的制备及光催化活性[J]. 化学学报, 2006, 64(2): 145-150.JIANG Hong-quan, WANG Peng, XIAN Heng-ze. Preparation and photocatalytic activities of low amount Yb3+-doped TiO2composite nano-powders[J]. Acta Chimica Sinica, 2006, 64(2):145-150.
猜你喜欢
建材发展导向(2021年16期)2021-10-12 05:39:24
陶瓷学报(2021年3期)2021-07-22 01:05:06
生态环境学报(2021年2期)2021-04-12 09:21:46
矿产综合利用(2020年1期)2020-07-24 08:51:04
云南化工(2020年2期)2020-02-18 06:24:02
新世纪智能(数学备考)(2019年9期)2019-10-16 11:44:58
湿法冶金(2019年4期)2019-08-08 08:43:54
中国锰业(2019年3期)2019-07-11 03:02:46
物理学进展(2017年1期)2017-02-23 01:35:44
大学化学(2015年5期)2015-09-18 08:43:48
