
直接甲醇燃料电池稳定性的初步研究
2012-07-05 01:39刘桂成蒋钜明王新东
电池 2012年5期
刘桂成,姜 颖,蒋钜明,王新东
(1.北京科技大学高效钢铁冶金国家重点实验室,北京 100083;2.北京科技大学物理化学系,北京 100083)
运行寿命是制约直接甲醇燃料电池(DMFC)商品化的主要因素之一。DMFC的内部工作环境较氢氧燃料电池复杂得多,运行条件更加苛刻。DMFC的性能衰减速率较高,数百、数千小时连续放电,可造成幅度达7%~50%的性能衰减[1-3],原因一般认为有:①DMFC的阴、阳极供料分别为空气(或氧气)和甲醇溶液,气/液不对称进料,使膜电极组件(MEA)承受较大的内应力,易变形;②为了应对甲醇渗透带来的负面影响,以及DMFC阳极甲醇氧化反应具有较高的过电位,通常DMFC催化剂的贵金属载量较高,因而造成催化剂粒子更易于聚集;③依据双功能机理,一般以PtRu为DM-FC的阳极催化剂,其中金属Ru在电池长期运行过程中的流失明显。
上述因素导致的DMFC稳定性过程运行中MEA的变化,具体表现为以下3种:
①催化剂失活,催化剂平均粒径的增大。Pt粒子以面心立方晶格存在,晶格参数为(220)的Pt晶面不受碳载体及Nafion聚合物衍射峰的干扰,可用来计算平均粒径。陈维民等[1]将DMFC在75℃下以70 mA/cm2的电流密度连续放电200 h,发现阳极催化剂颗粒的平均粒径几乎没有改变,阴极催化剂的平均粒径由5.8 nm增大至6.7 nm,阴阳极催化层的电化学表面积均显著减小,减小程度大于因催化剂颗粒长大带来的表面积损失。Z.B.Wang等[2]在60℃下将DMFC连续运行1 002 h,测得阴、阳极催化剂的平均粒径增大率分别为26.5%和18.5%。J.Guo等[3]在75℃下将DMFC连续运行2 020 h,发现阴、阳极催化剂的平均粒径增大率分别为90.9%和55.6%。现有报道表明:催化剂粒径在寿命测试过程中均有所增长,其中阴极催化剂粒径增长程度较阳极更大,原因一般认为是:阳极催化剂中 Ru的存在,对 Pt微晶的聚集可能存在抑制作用[4],而阴极催化剂存在于液相中,金属粒子迁移所需的活化能较低,即液相环境促进了Pt催化剂颗粒的团聚[5]。
②聚合物电解质的降解。聚合物电解质降解是增大M EA接触电阻的重要因素,◦OH自由基对聚合物电解质主侧链的攻击是造成降解的主要原因。通过 Raman光谱和FTIR光谱检测,发现:Nafion膜表面碳氟链与磺酸基的强度比发生了改变,体现了聚合物电解质的降解现象;且XPS结果显示,聚合物电解质的降解速率随温度和Fe3+浓度的升高而增大[6]。Z.Siroma等[7]发现:甲醇水溶液对 Nafion电解质具有一定的溶解作用,且溶解速率随温度和甲醇浓度的升高而增大,因此使用液体甲醇进料时,必须考虑甲醇溶液对聚合物电解质的溶解作用;同时,应避免杂质离子,如NH4+等,尤其是高价态的正离子,如 Ca2+、Fe3+等,即避免质子交换膜阳离子效应的出现。
③M EA微结构的老化。PTFE加入阴极扩散层,可改善气相传质能力,因而得到较广泛的使用。L.S.Sarma等[8]用能量色散X射线光谱仪(EDX)检测600 h放电运行后的阴极扩散层,发现F与C的物质的量比降低到初始值的58%,说明存在PTFE的降解。PTFE的降解会降低阴极扩散层的疏水性,使水分更易阻塞扩散层的微孔气体通道,增大了气相传质阻力,因此电池长期运行后会造成阴极水淹。
人们分析了催化剂、膜降解及MEA结构对电池稳定性的影响,但在电化学分析中未给出实时监测结果,也未明确阴阳极性能变化对MEA性能衰减的主次因素。本文作者在恒电位模式下进行了电池稳定性测试,采用三电极技术对阴阳极电位进行了实时监测,并结合电化学阻抗谱(EIS)及等效电路分析手段,对该测试过程中M EA性能的衰减机理进行了解析,同时从MEA关键材料的结构变化研究了阴阳极在MEA性能衰减方面各自不同的影响。
1 实验
1.1 MEA的制备与活化
以Vulcan XC-72导电炭黑(Cabot公司)和疏水处理后的TGP-H-090型碳纸(日本产)为主要材料,制备扩散层[9]。以PtRu黑(3 mg/cm2,英国产,Pt、Ru物质的量比为 1∶1,92%)、Pt黑(2 mg/cm2,英国产,95%)和Nafion 115(Du Pont公司)为主要材质,分别制备阳、阴极催化层。按文献[9-11]的方法进行质子交换膜的预处理,扩散层、阴阳极催化层的制备及MEA的立体化处理。将载有催化层的转移介质置于 Nafion 115膜的两侧,以 0.6~0.7 MPa的压力在135℃下热压150 s,再将扩散层以相同的条件热压至阴、阳极催化层的外侧,制成MEA,并对 MEA进行多层次活化[12]。
1.2 单体电池的稳定性及MEA性能的电化学测试
采用电位阶跃法,在VMP2型电化学综合测试系统(美国产)上评价MEA的性能。以饱和甘汞电极为参比电极,温度为 55℃,反应物为1.5 mol/L甲醇(国药集团,99.5%)溶液(2.5 ml/min)和氧气(670 ml/min),电位阶跃幅度为30 mV,每一步阶跃持续60 s,以达到电流的稳定,每次稳态极化曲线数据的采集均结束于短路电流。控制电池槽电位为0.4 V(vs.SHE,下同),测试电池的稳定性,运行条件同上。
在VMP2型电化学综合测试系统上进行EIS测试,仪器的工作电极与DMFC的阳极连接,参比电极及对电极与阴极连接。从高频到低频自动扫描,频率为10 mHz~100 kHz,交流信号正弦波振幅为20 mV,工作电位为0.4 V。
2 结果与讨论
2.1 电池在恒电位模式下的稳定性测试
将电池在0.4 V的恒电位模式下进行90 h的寿命测试,并检测运行过程中的阳极电位,结果见图1。

图1 DMFC在0.4 V恒电位模式下稳定运行的曲线Fig.1 Curves of stable working of DMFC at the constant potential mode of 0.4 V
从图1可知,电池的功率密度从初始值的52.2 mW/cm2衰减至2.8 mW/cm2,衰减率为 94.63%;阴、阳极电位均随着运行时间的延长而提高,阳极极化电位从0.502 V升高至0.547 V。在0.4 V恒电位的模式下,随着运行时间的延长,电池的性能衰减,即电流密度逐渐减小;随着电流密度的减小,阴极过电位减小、阳极过电位增大,因此电池性能的衰减主要来自阳极。90 h寿命测试前后电池的极化曲线见图2。

图2 90 h寿命测试前后MEA的极化曲线Fig.2 Polarization curvesof MEA before and after 90 h life test
从图2可知,在槽电位为0.4 V时,MEA的电流密度从开始的87.3 mA/cm2衰减至15.5 mA/cm2;当电流密度为15.5 mA/cm2时,寿命测试后,阳极过电位升高了0.112 V,阴极过电位仅升高了0.010 V。MEA的性能衰减,主要因素是阳极性能的损失。为进一步分析电池的稳定性与MEA性能衰减机理,进行了电池电压为0.4 V的交流阻抗测试。
2.2 采用EIS法解析电池稳定性运行中的性能衰减机理
在0.4 V的电池电压下,每10 h检测一次交流阻抗,结果见图3。

图3 电池稳定性测试中MEA在0.4 V下的电化学阻抗谱Fig.3 EIS of MEA at 0.4 V during the lifet test of DMFC
从图3b的高频端可见,电池内阻在初始阶段增大缓慢,随着运行时间的延长,内阻增大的速率越来越快,由初始的0.209 Ω◦cm2增大到 0.644 Ω◦cm2;在图 3a中频段,EIS 半圆弧的半径逐渐增大,即随着电池的运行,MEA的容抗逐渐增大,电化学反应的法拉第阻抗逐渐增大;而图3a中的低频端,随着电池的运行,甚至出现了浓差阻抗,可以推断,电池运行到80 h以后,MEA的结构瓦解,传质通道堵塞。
为了定量分析EIS数据,采用等效电路L1R1(CR2)[QR3(L2R4)][13-14]进行模拟,各元件参数的变化见图4。
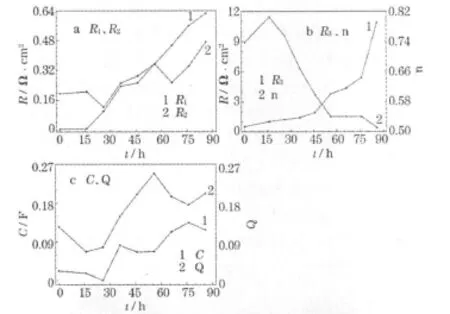
图4 电池稳定性测试中,EIS等效电路中各参数的变化Fig.4 Parameters variations of the equivalent circuit during lifet test of DMFC
从图4可知,阳极法拉第阻抗(R3)从0.524 Ω◦cm2增大至 10.880 Ω◦cm2,变化显著,尤其在最后20 h时,增大的速率骤增,表明此时阳极结构可能已经瓦解;电池的内阻(R1)由初始的 0.196 Ω◦cm2增大至 0.626 Ω◦cm2,可能是由于热压到催化层上的扩散层随着电池的运行被剥离。阴极电容C和阳极常相位角Q表现的电容性质随着电池的运行逐渐增大,且增幅相似,可能是阴、阳极催化层与质子交换膜之间产生的细缝所致。n值体现了阳极的传质特性,当n为0.5时,代表阳极出现浓差极化,在运行的前56 h内,n由0.737减小至0.511,阳极物料传质逐渐不畅,导致最后30 h内,一直保持浓差极化状态,限制了电池的性能。
2.3 MEA性能衰减机理的验证
为了验证对电池性能衰减因素的推论,拍摄了照片,并进行了SEM分析。经过稳定性测试后,扩散层和催化层及接触点处的变化见图5。

图5 电池稳定性测试对扩散层与催化层接触点的影响Fig.5 Effects of the stability test of DMFC on the contact points between diffusion layer and catalyst layer
从图5可知,由于流场板(c和f)的挤压,接触点上微孔层的碳粉脱落到催化层的表面。寿命测试后,阴极侧扩散层与催化层的接触极少,而阳极侧催化层和扩散层的接触相对较大。
这是由于阳极处于溶液环境,催化层随着质子交换膜一起溶胀,而扩散层基本无溶胀,造成扩散层和催化层的剥离,加之生成物CO2气体的冲刷,造成阳极微孔层中碳粉脱落较阴极微孔层严重;阴极侧随着电流密度的衰减,造成阴极侧产物水生成量减少,扩散层与催化层处于湿润的气体环境,催化层溶胀度较小,因此阴极微孔层有较少量的碳粉脱落。该现象证明:扩散层和催化层的剥离及微孔层中碳粉的脱落,导致了电池的内阻增大。
经过电池稳定性测试后的MEA的SEM图见图6。
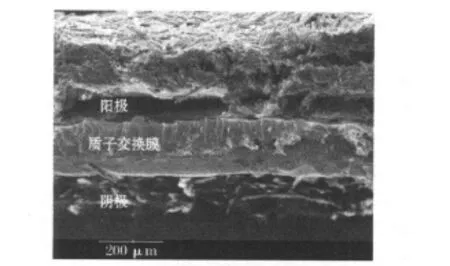
图6 经过电池稳定性测试后的MEA的SEM图Fig.6 SEM photograph of M EA after the stability test of DMFC
从图6可知,阴极的微观结构保持得比较好,而阳极的微观结构中,出现了扩散层与催化层的剥离,以及催化层的瓦解。
这主要是因为热压工艺制备的阳极扩散层在溶液环境中,微孔层首先被剥离,扩散层和催化层开裂,进而导致阳极物料传质困难和电池内阻的增大。产物气体的冲刷,长期积累导致阳极催化层结构瓦解,表现在电化学反应中,即为阳极法拉第阻抗增大。
3 结论
采用0.4 V的恒电位模式进行DMFC稳定性测试,同时以三电极体系检测寿命测试过程中的阳极电位,结合寿命测试前后单体电池极化曲线的变化,可得出电池稳定性测试过程中,MEA性能衰减主要来自阳极。
通过交流阻抗谱及等效电路分析,结合MEA结构变化的照片和SEM图的对比,得出了MEA性能衰减的机理主要是:①由于MEA微结构的变化,导致电池的内阻逐渐增大,主要表现为阳极扩散层的剥离以及微孔层中碳粉的脱落;②阳极催化层结构的瓦解,导致了阳极电化学反应法拉第阻抗的增大。
延长DMFC稳定性的措施,可以从进一步改进MEA的制备工艺、优化单体电池的工作模式以及操作参数等方面进行考虑。
[1]CHEN Wei-min(陈维民),SUN Gong-quan(孙公权),ZHAO Xin-sheng(赵新生),et al.直接甲醇燃料电池电催化剂性能衰减研究[J].Chemical Journal of Chinese Universities(高等学校化学学报),2007,28(5):928-931.
[2]Wang Z B,Rivera H,Wang X P,et al.Catalyst failure analysis of a direct methanol fuel cell membrane electrode assembly[J].J Power Sources,2008,177(2):386-392.
[3]Guo J,Sun G,Wu Z,et al.The durability of polyol-synthesized PtRu/C for direct methanol fuel cells[J].J Power Sources,2007,172(2):666-675.
[4]Antolini E,Giorgi L,Cardellini F,et al.Physical and morphological characteristics and electrochemical behavior in PEM fuel cells of PtRu/C catalysts[J].J J Solid State Electrochem,2001,5(2):131-140.
[5]Bett J A S,Kinoshita K,Stonehart P.Crystallite growth of platinum dispersed on graphitized carbon black(Ⅱ).Effect of liquid environment[J].J Catal,1976,41(1):124-133.
[6]Chen C,Levitin G,Hess D W,et al.XPS investigation of Nafion○Rmembrane degradation[J].J PowerSources,2007,169(2):288-295.
[7]Siroma Z,Fujiwara N,Ioroi T,et al.Dissolution of Nafion○Rmembrane and recast Nafion○Rfilm in mixtures of methanol and water[J].J Power Sources,2004,126(1-2):41-45.
[8]Sarma L S,Chen C H,Wang G R,et al.Investigations of direct methanol fuel cell(DMFC)fading mechanisms[J].J Power Sources,2007,167(2):358-365.
[9]LIU Gui-cheng(刘桂成),ZHANG Hao(张浩),WANG Yi-tuo(王一拓),et al.液体进料直接甲醇燃料电池工况的匹配及优化[J].Battery Bimonthly(电池),2012,42(1):7-10.
[10]Liu G C,Wang M,Wang Y T,et al.Anode catalyst layer with novel microstructure for a direct methanol fuel cell[J].Int J Hydrogen Energy,2012,37(10):8 659-8 663.
[11]LIU Gui-cheng(刘桂成),WANG Yi-tuo(王一拓),WANG Meng(王萌),et al.DM FC用膜电极组件的结构及性能被动式DMFC的性能[J].Battery Bimonthly(电池),2012,42(2):66-69.
[12]Liu G C,Xu J Y,Wang T T,et al.The performance and mechanism of multi-step activation of MEA for DMFC[J].Int J Hydrogen Energy,2010,35(22):12 341-12 345.
[13]Piela P,Fields R,Zelenay P.Electrochemical impedance spectroscopy for direct methanol fuel cell diagnostics[J].J Electrochem Soc,2006,153(10):A1 902-A1 913.
[14]Du C Y,Zhao T S,Yang W W.Effect of methanol crossover on the cathode behavior of a DMFC:a half-cell investigation[J].Electrochim Acta,2007,52(16):5 266-5 271.
猜你喜欢
化工管理(2022年14期)2022-12-02
陶瓷学报(2021年1期)2021-04-13
电子制作(2018年12期)2018-08-01
材料科学与工程学报(2016年1期)2017-01-15
光学精密工程(2016年4期)2016-11-07
中国塑料(2016年4期)2016-06-27
船舶标准化工程师(2015年5期)2015-12-03
中国塑料(2015年3期)2015-11-27
中国塑料(2015年7期)2015-10-14
