
高场非对称波形离子迁移率谱的正弦切割波形产生与离子分离特性*
2012-06-10 08:08赵东杰高晓光何秀丽李建平
传感技术学报 2012年7期
赵东杰,贾 建,高晓光,何秀丽,李建平
(中国科学院电子学研究所,传感技术联合国家重点实验室,北京100190)
近年来出现的高场非对称波形离子迁移率谱仪(high-field asymmetricion mobilityspectrometer,FAIMS),具有结构简单、尺寸小、分辨率高、易于与色谱、质谱联用等特点,在挥发性有机化合物、爆炸物、化学战剂、蛋白质等物质检测方面已有应用研究,受到了越来越多的关注[1-2]。
FAIMS通过在两个电极(平板电极或同轴圆柱电极)之间形成高低不对称电场,利用离子在高低电场下离子迁移率的非线性变化实现气相离子分离。FAIMS中实现高低不对称电场时需要高压(Vp-p>1 000 V)、高频(~1 MHz)非对称电压波形(Asymmetric Waveform Voltage,AWV)。理想的非对称波形是矩形波[3],可通过高压高速开关电路切换正负高压电源实现,但是在工程实现上难度较大,装置体积较大且功耗高[4]。因此出现了其它类型的非对称波电压波形[5],例如正弦叠加波形,由一个正弦波与其相移90度的二次谐波叠加而成,通常由两个振荡电路输出波形叠加实现,但存在频率易漂移波动的问题;还有利用变压器反激升压电路生成的正弦切割波,其波形介于矩形波形与正弦叠加波之间,可以看成由正弦波正半部分与矩形波负半部分组合而成,实现电路具有体积小、效率高等特点,但非对称波形的高频高压对变压器制备要求较高[6]。因此,研究低成本、简易的正弦切割波发生器制备方法,进而研究所产生波形对FAIMS的影响,由此来优化工作参数,有利于改善FAIMS分离效果,并实现FAIMS系统低成本化及微型化。
本文制备了一种基于电感反激电路的高压高频正弦切割电压波形发生器,分析了该正弦切割波形的变化特点,将波形应用于紫外光电离化FAIMS器件,对低浓度的有机挥发物(VOC)丙酮、环己烷和乙醇样品进行实验,研究波形对图谱离子峰位及分辨率的影响。
1 实验部分
1.1 FAIMS 实验装置
采用自制的平板型FAIMS装置进行实验,结构如图1所示,离化源为10.6 eV紫外灯,正离子检测模式。利用射频溅射镀膜技术在玻璃基片上制备出过滤电极(尺寸为11 mm×5 mm),检测电极以及偏转电极(尺寸均为5 mm×5 mm)。上下两片玻璃基片由0.35 mm玻璃压条隔开,形成用于通过样品气体的狭缝。非对称波形AWV与补偿电压(Compensated Voltage,CV)叠加在过滤电极上,补偿电压为周期为1 s的锯齿波,幅度从-21 V至+21 V。偏转电极上的偏转电压(Deflected Voltage,DV)为+12 V。高纯氮气作为载气,由质量流量计控制流量。离子信号采用自制的精度为0.1 pA的电流信号检测电路,并由Tektronix TDS2002型示波器记录波形。

图1 FAIMS结构原理图
1.2 正弦切割波形发生器
本文采用电感与高压MOSFET管组成正弦切割波发生器电路,如图2所示。当MOSFET管开启时,电感L1进行储能;MOSFET管关断时,由于电感电流不能突变,电感储能释放在GB点形成高压脉冲,波形类似正弦波的正半周期。MOSFET管栅极输入方波开关信号,使MOSFET管周期性开关,则形成周期脉冲,通过瓷片电容C1滤除波形中的直流分量后形成正弦切割波AWV。该波形与补偿电压CV耦合,输出到FAIMS器件。图中的电感L2将高压高频的AWV与CV产生电路相隔离,同时又保证低频低压CV信号通过并叠加在AWV信号上。此外在输出回路并联电容C2、C3组成的分压电路用于观察输出波形。通过调节MOSFET栅极上的开关信号频率(如采用标准信号源),使输出正弦切割波频率在0.8 MHz~1.5 MHz范围内精确可调;通过调节DC端的直流驱动电压,使输出电压波形峰峰值在0~1 500 V范围连续可调;调整电感L1的大小可以改变正周期脉冲的宽度。通过测量,电路功耗在10 W以下,保证了长期的工作稳定性。
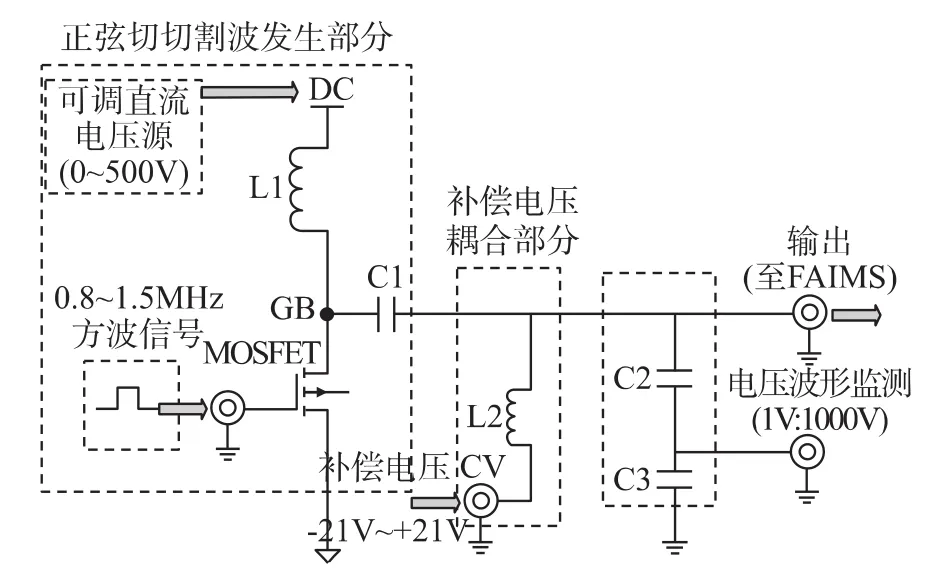
图2 正弦切割波发生电路原理框图
1.3 样品制备
使用有机挥发物(VOC)丙酮、环己烷和乙醇作为检测对象,实验样品气体采用扩散原理制备[7-9]。分别将0.2 mL的分析纯级丙酮、环己烷、乙醇样品装入1.5 mL的色谱样品瓶内密封,将不锈钢针管插入瓶内,样品将通过针管扩散出来。将扩散瓶放入1 000 mL锥形瓶中,利用水浴保持恒温,锥形瓶内通入空气稀释形成样品气体。通过减重法测量丙酮、环己烷和乙醇样品浓度分别为:10 μg/L、13 μg/L 和7 μg/L。
2 结果与讨论
2.1 正弦切割波发生器的输出波形
用于FAIMS的非对称波形需要满足两个条件:(1)波形函数F(t)的单周期积分为0,即正负周期波形的面积相等,见式(1);(2)波形函数F(t)奇数次积分不为0,即保证波形的非对称性,见式(2)。

通常用式(2)来衡量波形非对称性,称为波形因数(Form Factor)[3,10]。
在0.8 MHz~1.4 MHz范围内设定不同频率,以及调节波形的正弦部分最大幅度(设为波形幅值VD)从0 V到1 000 V,分别观测输出波形的变化。
图3(a)是VD设定在800 V,在0.8 MHz~1.4 MHz范围各频率下的输出波形。观察到在不同频率下,输出波形的正周期正弦部分保持不变,但负周期部分的时间宽度随着频率的增加而减小。图3(b)是频率固定在1.16 MHz时不同VD的波形,图中可见波形的占空比基本不变,不随幅值改变。
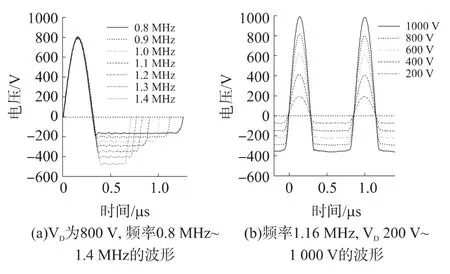
图3 波形发生器输出的非对称波形
2.2 正弦切割波频率对离子分离效果的影响
固定波形幅值 VD,设定频率从 0.8 MHz~1.4 MHz,分别通入配置出的丙酮、环己烷和乙醇样品气体,测量各自CV扫描谱,见图4。在图4中,丙酮与环己烷在非对称分离电场的作用下出现两个离子特征峰,分别用P1和P2表示,其中P1峰位随着频率变化较大,而P2的峰位相对保持稳定。根据相关文献报道[11-12]的检测结果,可以确定三种物质的P1峰均为其单体特征离子峰,而丙酮与环己烷的P2峰为其二聚体特征峰。单体特征峰位置随波形频率有明显变化,说明其离子迁移率对非对称波形变化较敏感。
对于FAIMS,离子分离效果反映为离子峰位和分辨率变化。离子峰位值是离子峰位置与原点(0V)的距离,峰位值大说明被检测物质的离子迁移率变化大,更适合于FAIMS检测;而分辨率用峰位除以半高峰宽(FWHM)来表示[3]。

其中R为检测分辨率,A为离子峰峰位值(V),W为特征离子峰的FWHM(V),反映了离子峰的尖锐程度,R的值越大说明分辨率越好。
图5(a)是由图4得到的单体离子峰(P1)的峰位值变化曲线。丙酮与环己烷的峰位变化趋势基本一致,在0.8 MHz至1 MHz之间,随着频率的增加,峰位值变大,而1 MHz之后,离子峰位值变小;乙醇在 0.8 MHz~1.2 MHz之间峰位变化不明显,在1.2 MHz以后,峰位值变小。
图5(b)是分辨率变化曲线,三种物质的检测分辨率都呈现先升后降的趋势,在1 MHz处分辨率最好;在1.4 MHz处分辨率最差。

图5 样品气的特征峰的峰位(a)、分辨率(b)随频率的变化
频率的改变使得正弦切割波的负周期波形发生较大变化,因此观测各频率下波形的波形因数及占空比γsin(正弦部分与整个波形周期的比值)变化,见表1。波形因数变化趋势与测量得到的分辨率变化(图5b)基本保持一致。如在波形因数取值最大的频率点1 MHz(γsin=0.42),分辨率最好,说明此时波形非对称性最好;而在频率1.4 MHz,其波形因数值最小,分辨率值最低,非对称性也最差。

表1 0.8 MHz~1.4 MHz正弦切割波的波形因数
2.3 正弦切割波幅值对离子分离效果的影响
将频率定为1 MHz,调节VD由0至1 000 V,得到样品气的不同电场强度下的CV扫描强度谱,观察到随着电场强度的增加,离子峰峰位偏移变大,同时离子峰强度下降。
图6为三种物质的峰位和分辨率随VD幅值的变化趋势图。图6(a)中三种物质的峰位均随VD增加而增加:其中当VD在400 V以下时,产生的非对称电场不足以使离子峰发生偏移,离子迁移率为常值,对离子没有分离作用;在VD大于600 V之后,峰位随电场强度的增大而有明显增加。图6(b)中三种物质的分辨率也随非对称波形电压的升高而增加:VD大于400 V后,分辨率随VD的增加而提高,即高场强下,离子迁移率差值随非对称波形电场强度的增加而不断增大,分辨率也随之升高。

图6 波形幅值VD对样品气的峰位(a)、分辨率(b)的影响
3 结论
FAIMS高压高频不对称波形的产生是制约其应用的瓶颈。本文提出一种单电感反激电路产生正弦切割波形的方法,简化了电路结构,降低了功率。使用紫外光离化的平板型FAIMS,研究了不同频率与幅度的正弦切割波对有机挥发物(丙酮、环己烷和乙醇)的离子分离效果的影响。在正半周期输出幅值固定,正弦切割波形在占空比为0.42时,波形因数最高,被检测物质的离子峰位和分辨率最大;在固定频率时,FAIMS的被测物离子分辨率随不对称电场强度的增强而增加。
[1]Kolakowski B M,Mester Z.Review of Applications of High-Field Asymmetric Waveform Ion Mobility Spectrometry(FAIMS)and Differential Mobility Spectrometry(DMS)[J].Analyst,2007,132:842-864.
[2]Shvartsburg A A,Tang K Q,Smith R D.Differential Ion Mobility Separations of Peptides with Resolving Power Exceeding 50[J].Analytical Chemistry,2010,82:32-35.
[3]Shvartsburg A A,Smith R D.Optimum Waveforms for Differential Ion Mobility Spectrometry(FAIMS)[J].Journal of the American Society for Mass Spectrometry,2008,19:1286-1295.
[4]Papanastasiou D,Wollnik H,Rico G,et al.Differential Mobility Separation of Ions Using a Rectangular Asymmetric Waveform[J].Journal of Physical Chemistry A,2008,112:3638-3645.
[5]Krylov E V,Coy S L,Vandermey J,et al.Selection and Generation of Waveforms for Differential Mobility Spectrometry[J].Rev Sci Instrum,2010,81:24101.
[6]Krylov E V,Pulses of Special Shapes Formed on a Capacitive Load[J].Instruments and Experimental Techniques,1997,40:628-631.
[7]Arce L,Menendez M,Garrido-Delgado R,et al.Sample-Introduction Systems Coupled to Ion-Mobility Spectrometry Equipment for Determining Compounds Present in Gaseous,Liquid and Solid Samples[J].Trac-Trends in Analytical Chemistry,2008,27:139-150.
[8]王乐,王镝,於锦,等.基于谐振型SAW传感器的呼吸检测系统设计[J].传感技术学报,2011,24(4):498-502.
[9]郭希山,童基均,陈裕泉,等.用于室内有毒气体快速检测的便携式 CC/SAW 电子鼻[J].传感技术学报,2006,19(1):68-73.
[10]Krylov E V,Nazarov E G,Miller R A.Differential Mobility Spectrometer:Model of Operation[J].International Journal of Mass Spectrometry,2007,266:76-85.
[11]Krylov E,Nazarov E G,Miller R A,et al.Field Dependence of Mobilities for Gas-Phase-Protonated Monomers and Proton-Bound Dimers of Ketones by Planar Field Asymmetric Waveform Ion Mobility Spectrometer(PFAIMS)[J].Journal of Physical Chemistry A,2002,106:5437-5444.
[12]Nazarov E G,Miller R A,Eiceman G A,et al.Miniature Differential Mobility Spectrometry Using Atmospheric Pressure Photoionization[J].Analytical Chemistry,2006,78:4553-4563.
猜你喜欢
化工管理(2021年7期)2021-05-13
同位素(2020年6期)2020-12-18
人民交通(2020年17期)2020-09-15
环境保护与循环经济(2020年4期)2020-06-08
航天电子对抗(2019年4期)2019-06-02
中国特种设备安全(2019年1期)2019-03-13
数学年刊A辑(中文版)(2018年2期)2019-01-08
光谱学与光谱分析(2016年10期)2016-07-12
电测与仪表(2015年4期)2015-04-12
无机化学学报(2014年3期)2014-02-28
