
基于分子对接研究吲哚咔唑类小分子对Tie-2/VEGFR2的双效抑制作用模式*
2012-05-09 08:12田元新张贤祚安林坤
中山大学学报(自然科学版)(中英文) 2012年2期
田元新,张贤祚, 安林坤
(1.南方医科大学药学院,广东 广州510515;2.南方医科大学第一临床医学院,广东 广州510515;3.中山大学药学院,广东 广州510006)
抗血管生成疗法——阻断实体瘤血管内皮增殖这一关键步骤,一直是药物开发领域的热点。血管的发生和生成对于早期胚胎发育以及包括肿瘤、糖尿病、风湿性关节炎等的一系列疾病过程至关重要[1],其确切的调节机制尚未被研究清楚。目前通常认为与血管生成过程密切相关的有血管内皮生长因子VEGF及其受体系统和血管生成素及其受体酪氨酸激酶Tie-2系统。VEGF及其受体VEGFR(主要是VEGFR2)可介导中胚层细胞向内皮细胞分化并移行至血管内皮处;而血管生成素系统则与血管生成晚期血管重构、成熟过程中的细胞间相互作用、细胞与基质间的相互作用有关[2-3]。因此,有效地一并阻断两者可能收获比单一阻断疗法更满意的疗效[4-5]。基于CEP-7055设计的一系列吲哚咔唑类化合物对VEGFR2和Tie-2均有抑制作用,目前已有药物处于临床应用前阶段,部分处于临床活性评价阶段[6-10]。该类Tie-2/VEGFR2双位点抑制剂的3D-QSAR和体外活性已被证实,但与两类受体结合机制的异同尚未明确。
本研究以现有多靶点酪氨酸激酶抑制剂吲哚咔唑类小分子[6-10],以及VEGFR2、Tie-2的结构为出发点[11],利用分子对接技术研究抑制剂分子与VEGFR2/Tie-2结合的复合三维结构模型[12-14],微观阐明它与受体蛋白Tie-2及VEGFR2的相互作用模式,通过研究他们和受体蛋白相互作用的异同,分析配体结构和与Tie-2、VEGFR2间结合活性差异的关系,为设计新型高效多靶点抑制剂化合物提供理论指导。
1 研究方法
1.1 小分子的构建
从Tie-2的蛋白质结晶体(蛋白质PDB结构数据库http:∥www.rcsb.org/pdb;ID:3L8P)中提取出配体CEP112071的构象,并以此构象为模板构建出共80个目标分子(见表1,表2)。添加Gasterger-Hückel电荷,采用TRIPOS力场,以共轭梯度法优化得到最低能量构象,收敛标准为0.001 kcal/(mol·A)。以此为起始构象,用SYBYL-7.3中进行分子对接研究[12,15]。
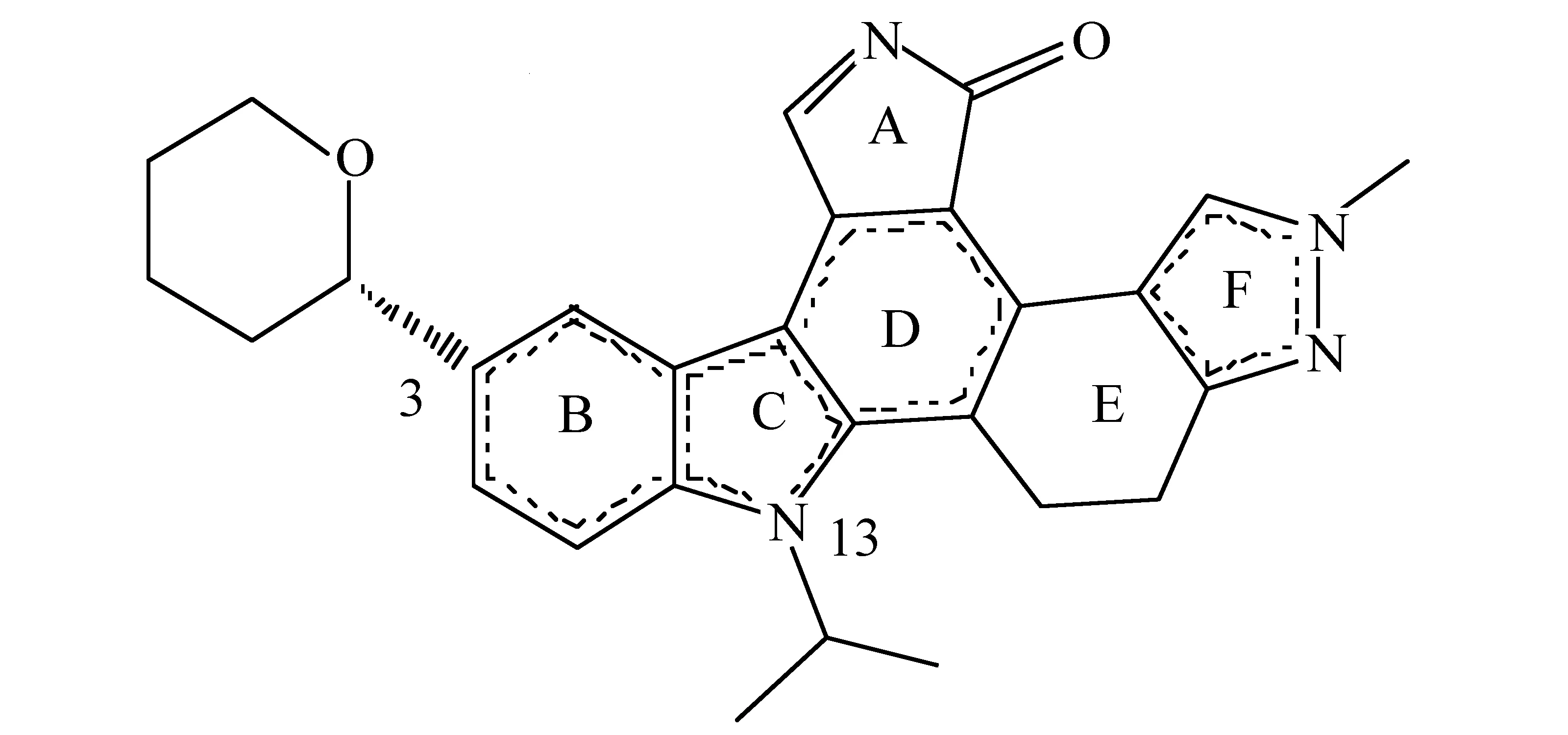
图1 CEP112071化合物的化学结构
1.2 分子对接
用于分子对接计算的VEGFR2和Tie-2蛋白质晶体结构取自蛋白质PDB结构数据库(VEGFR2:1YWN, Tie-2:3L8P)。对接前除去晶体结构中与受体结合的原配体分子和水分子,并对受体蛋白进行加氢并优化,后在配体引导下生成protomol原型分子。分子表面的产生用molcad模块。如无特别说明,所有参数均采用SYBYL-7.3默认值。Surflex-Dock打分函数以-log10为单位模拟结合能力,得分最高的构象被认为是最佳构象[16]。
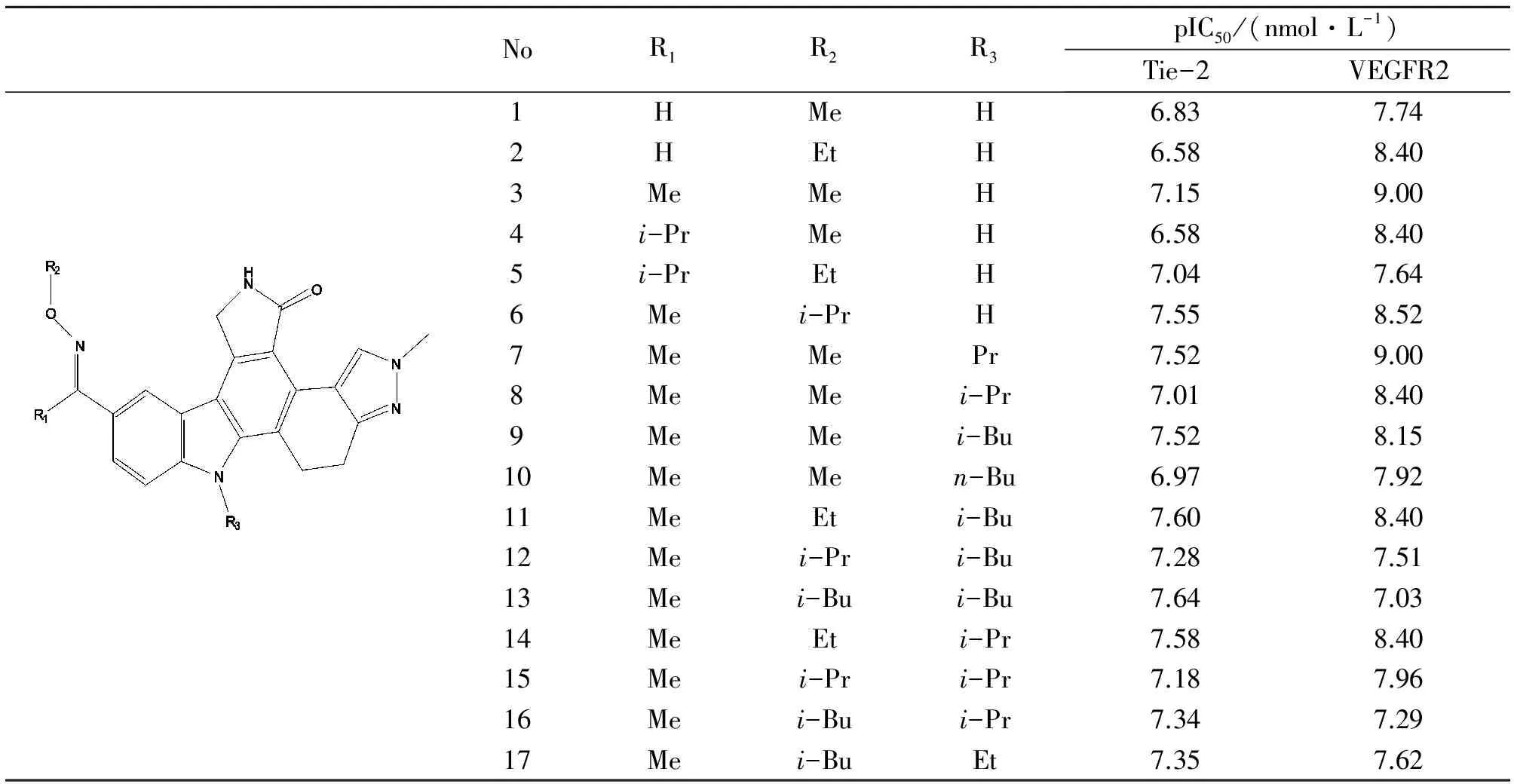
表1 1~17号双效抑制剂的分子结构与其生物活性[6-8,10]
表2 18~80号双效抑制剂的分子结构与其生物活性[9]
Table 2 Molecular structure of dual inhibitors No.18~80 and their biological activities[9]
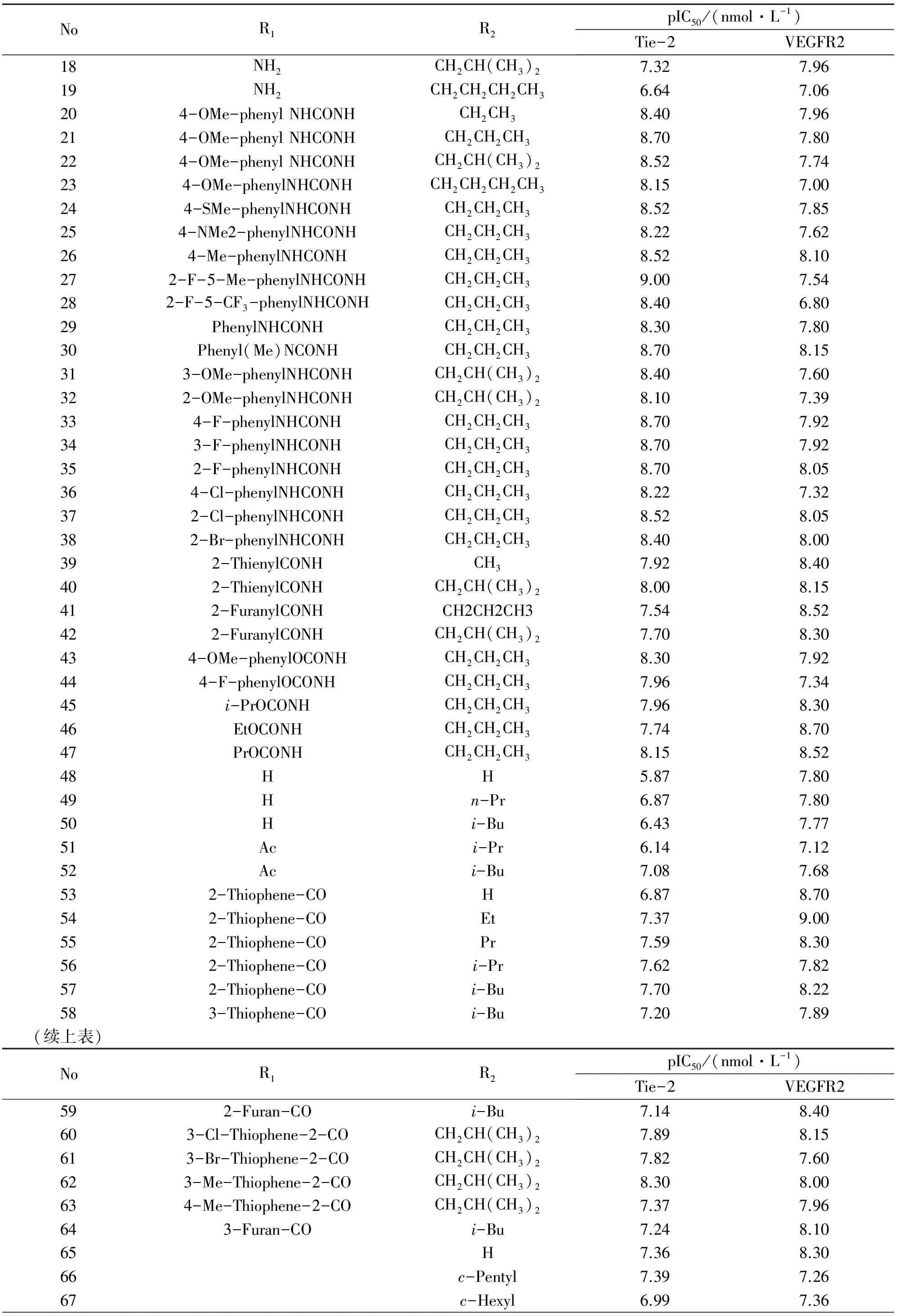
NoR1R2pIC50/(nmol·L-1)Tie-2VEGFR218NH2CH2CH(CH3)27.327.9619NH2CH2CH2CH2CH36.647.06204-OMe-phenylNHCONHCH2CH38.407.96214-OMe-phenylNHCONHCH2CH2CH38.707.80224-OMe-phenylNHCONHCH2CH(CH3)28.527.74234-OMe-phenylNHCONHCH2CH2CH2CH38.157.00244-SMe-phenylNHCONHCH2CH2CH38.527.85254-NMe2-phenylNHCONHCH2CH2CH38.227.62264-Me-phenylNHCONHCH2CH2CH38.528.10272-F-5-Me-phenylNHCONHCH2CH2CH39.007.54282-F-5-CF3-phenylNHCONHCH2CH2CH38.406.8029PhenylNHCONHCH2CH2CH38.307.8030Phenyl(Me)NCONHCH2CH2CH38.708.15313-OMe-phenylNHCONHCH2CH(CH3)28.407.60322-OMe-phenylNHCONHCH2CH(CH3)28.107.39334-F-phenylNHCONHCH2CH2CH38.707.92343-F-phenylNHCONHCH2CH2CH38.707.92352-F-phenylNHCONHCH2CH2CH38.708.05364-Cl-phenylNHCONHCH2CH2CH38.227.32372-Cl-phenylNHCONHCH2CH2CH38.528.05382-Br-phenylNHCONHCH2CH2CH38.408.00392-ThienylCONHCH37.928.40402-ThienylCONHCH2CH(CH3)28.008.15412-FuranylCONHCH2CH2CH37.548.52422-FuranylCONHCH2CH(CH3)27.708.30434-OMe-phenylOCONHCH2CH2CH38.307.92444-F-phenylOCONHCH2CH2CH37.967.3445i-PrOCONHCH2CH2CH37.968.3046EtOCONHCH2CH2CH37.748.7047PrOCONHCH2CH2CH38.158.5248HH5.877.8049Hn-Pr6.877.8050Hi-Bu6.437.7751Aci-Pr6.147.1252Aci-Bu7.087.68532-Thiophene-COH6.878.70542-Thiophene-COEt7.379.00552-Thiophene-COPr7.598.30562-Thiophene-COi-Pr7.627.82572-Thiophene-COi-Bu7.708.22583-Thiophene-COi-Bu7.207.89(续上表)NoR1R2pIC50/(nmol·L-1)Tie-2VEGFR2592-Furan-COi-Bu7.148.40603-Cl-Thiophene-2-COCH2CH(CH3)27.898.15613-Br-Thiophene-2-COCH2CH(CH3)27.827.60623-Me-Thiophene-2-COCH2CH(CH3)28.308.00634-Me-Thiophene-2-COCH2CH(CH3)27.377.96643-Furan-COi-Bu7.248.1065H7.368.3066c-Pentyl7.397.2667c-Hexyl6.997.36
2 结果与讨论
2.1 分子对接方法的确认
为验证对接方法的可信度,对接前分别将3L8P和1YWN复合物中的配体从受体的活性口袋中取出,优化后再对接回结合口袋。对接产生的构象与复合物中的构象十分接近,均方根位移(RMSD)分别为0.065 30和0.072 60 nm,低于实验的最小分辨率,说明对接方法得到的结果是可靠的。
将小分子数据集分别与VEGFR2、Tie-2进行对接,比较分子对接结果。研究表明,吲哚咔唑类分子对VEGFR2/Tie-2的双效抑制作用主要源于相似的结合模式:配体在两种受体结合位点处的活性口袋位置相似,且相似位置的氨基酸残基提供了氢键的受体或供体,形成螯合氢键,部分关键性的氨基酸残基表现出相似的静电特征,故吲哚咔唑类分子与两类受体均有良好的结合活性,表现出双效抑制剂的特征。然而,由于活性口袋的形状大小有所差异,分子间的相互作用力不同,配体分子在两类受体间的活性仍有差异,具体体现在下述几个方面。
2.2 疏水作用分析
相关参考研究的计算表明,VEGFR2受体活性结合位点大体含有5个区域:①与绞链区相邻的疏水残基Val846,Ala864,Val897,Val914,Phe916,Leu1033组成的一个扁平的区域为疏水腔Ⅰ,该疏水腔也是ATP的腺嘌呤环的结合位点,因此是抑制剂结合的关键区域。②由疏水残基Ile886,Leu887,Ile890,Val896,Val897,Leu1017,Ile1042组成疏水腔Ⅱ;疏水腔Ⅰ和Ⅱ之间有1个由Lys866,Glu883的侧链和Asp1044骨架上的羰基氧共同形成的环形的小极性区域。③脂肪族疏水基团(异丁烷、环乙烷)在活性腔开口向溶剂处的Leu838和Phe916的侧链附近也有分布。④在Arg1030残基附近由Leu1033和Cys1043组成的疏水区域Ⅳ。另外,苯环在Arg1030的胍基附近也有分布,形成阳离子-π相互作用[17-18]。
对Tie-2的受体活性口袋进行分析,根据其疏水性质将其中关键区域划分为两个区域:Ⅰ区由疏水残基Ile830,Val838,Ala853,Ile886,Leu971构成狭窄扁平的输水腔,Ⅱ 区由Val875,Leu876,Leu888,Leu900,Ile902,Phe983构成另一空间较小半封闭区域,Asp982的极性亲水残基在这里起到了隔断两个疏水腔的作用。

图2 结合位点疏水腔的划分以及疏水性质的差异
从活性位点亲水性和疏水性表面的分布(图2)发现,二羟吲哚咔唑分子母核均与活性腔Ⅰ区结合,3号C原子侧链疏水基团均结合于疏水腔Ⅱ。不同的是Tie-2的Ⅱ区域空间更小,疏水作用对整体活性影响更大,例如11号和12号分子仅在3号C原子侧链的顶端有微小不同,两者Tie-2活性相似而VEGFR2活性则差异较大。Tie-2没有对应于Ⅲ区的疏水区域,故对于配体分子13号N原子连接侧链区域的选择性较弱,对接中我们发现16号分子和17号分子在该区域分别为异丙基和乙基,两者的Tie-2活性相似而VEGFR2活性有所差异,说明此处是VEGFR2的关键选择性位点,Tie-2则不然。配体分子F环显亲水性,与Tie-2开口向溶剂处Gly908亲水区域结合,提高与Tie-2的结合亲和力,我们推测在这里引入侧链疏水基团,如烃基、烯烃基等,有助于提高配体的VEGFR2活性,而保留或加强亲水性基团、将杂氮环改为芳环,则有利于提高与Tie-2的结合活性。
2.3 静电作用分析
静电作用方面,配体分子侧链酰胺键两个N原子侧翼呈正电性、O呈负电性,与VEGFR2的对接中和Glu883间存在较强吸引,同时吸引突出于受体表面的Asp1044弯向Lys866,形成鳌状钳住配体分子;类似的,与Tie-2的对接中与Lys855形成吸引,诱使Lys855突出表面弯向负电性的Asp982,嵌住配体分子。两者不同之处在于:A环上显负电的杂氮原子以及以碳氧双键结合于环上的氧原子,可与VEGFR2的Phe916、Cys917互相吸引,有利于配体分子在VEGFR2活性口袋中相对位置的固定; F环上呈强负电性的杂氮原子,可与VEGFR2的Leu838相吸引,同时吸引Glu920的突出部分弯向Leu838,同样对配体分子起到固定作用。另一方面,我们发现13号N原子连接的侧链与VEGFR2对接时处于正电性的Glu839、Arg840附近,在与Tie-2对接时则靠近具有负电性质的Glu832。如果我们在该区域引入具有强电荷性质的基团,可能收获较理想的选择性(图3)。
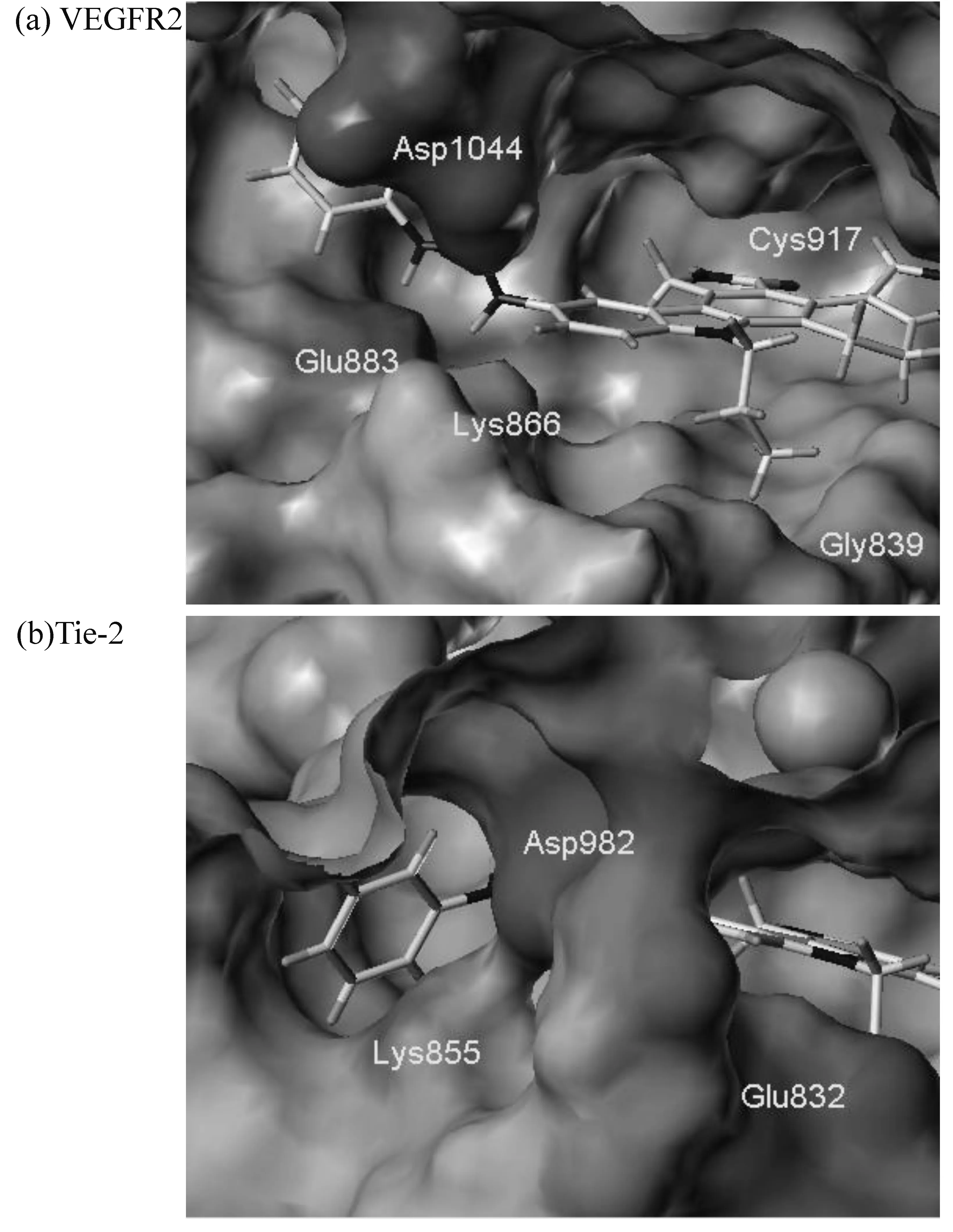
图3 结合位点表面静电性质的差异
2.4 氢键分析

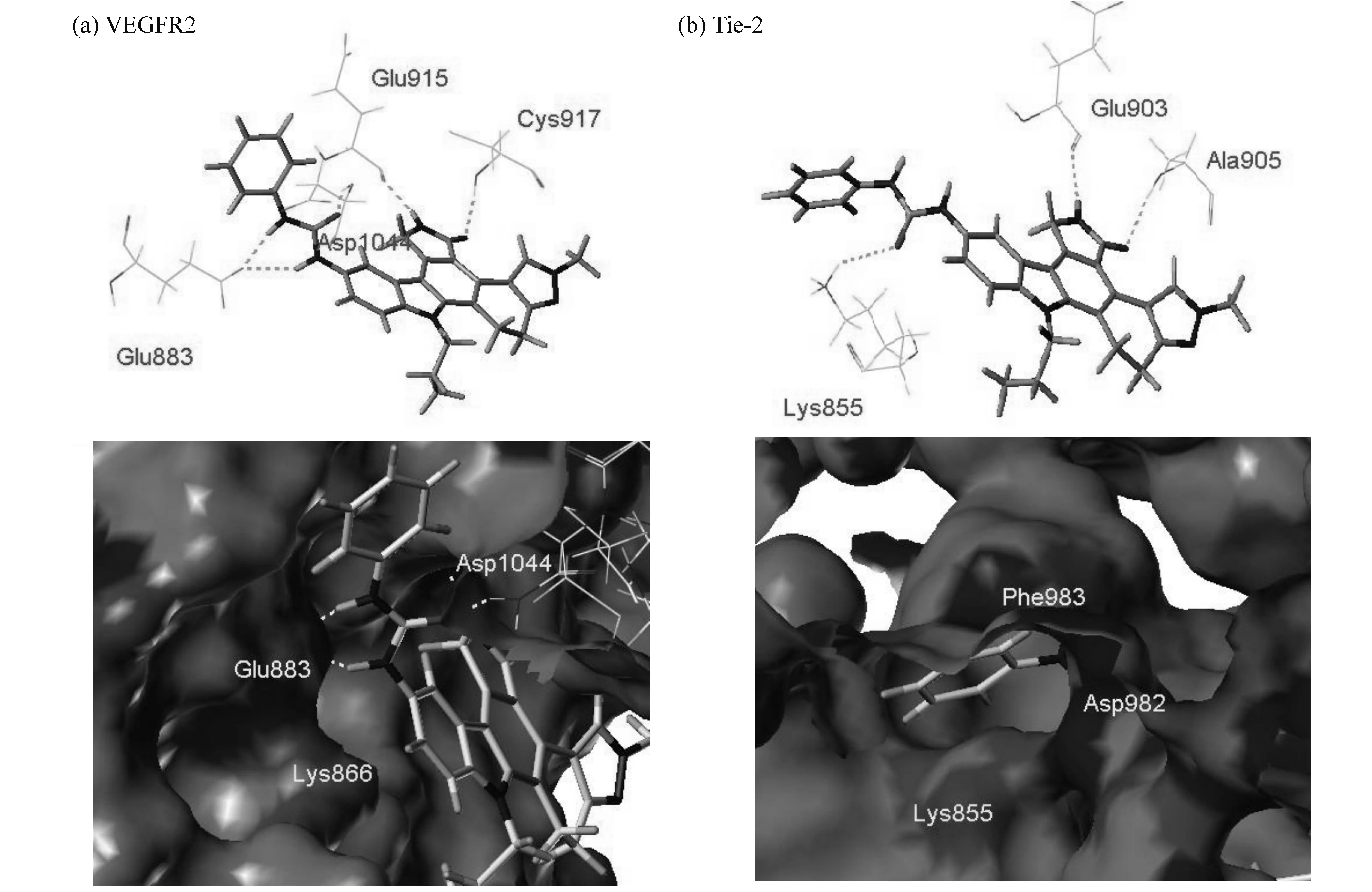
图4 VEGFR2/Tie-2与配体的结合模式
两者不同处在于3号C原子侧链附近,VEGFR2上Asp1044仅提供氢键供体,相应位置Tie-2的Phe983和Asp982则可分别提供潜在供、受体。在受体另一侧,Glu883和Lys866分别提供氢键受体和供体,而Tie-2相应位置则只有Lys855能提供氨基作为氢键供体。此处氢键供受体构象的翻转使得配体分子在结合时结合构象不同,对结合活性产生了影响。
2.5 空间位阻分析
在以上的对接分析中我们还发现,VEGFR2的活性位点较Tie-2的更为开放,未完全地包裹配体分子,使得由疏水力、静电作用和氢键作用带来的影响远不及Tie-2明显,例如23号分子在3号C原子侧链的顶端连有一个甲氧基,表现出良好的Tie-2活性,但对VEGFR2的活性较差。同时,更加开放的活性口袋也为配体分子的侧链伸展提供了可能性。如果我们在各个侧链上引入体积较大的基团,或是延长侧链,则可能降低候选分子与Tie-2的结合活性,达到特异性选择的目的。
3 结论与讨论
本文通过分子对接的方法对吲哚咔唑系列分子和VEGFR2/Tie-2的相互作用做了研究,分析了该系列分子与受体结合的最佳结构、构象和结合机制,分子对接模拟得到的结果与所揭示的机制与相关文献报道的结果相吻合。研究表明,抑制剂分子主要通过疏水作用、氢键和静电作用嵌合在激酶受体的活性位点中,使其催化活性受到抑制,干扰血管内皮的生长,从而对肿瘤的产生起到干预作用。其中,两类受体间表面疏水腔性质的不同是影响配体选择性的主要原因。本研究对阐明该系列分子的抗肿瘤机制具有理论指导作用,进而为药物分子设计提供了理论依据。
本研究的结果说明,不同激酶受体之间可能存在着相同或者相似的结构区域,与特定配体的结合特性相近,可以通过一系列配体化合物达到抑制多个靶点的效果。分析不同激酶受体在结合模式上的不同有助于设计具有高效选择性的抑制剂药物,在保证药效的情况下提高药物的针对性降低毒副作用,使设计药物具有临床实用的可能。
参考文献:
[1]CRIVELLATO E.The role of angiogenic growth factors in organogenesis[J].International Journal of Developmental Biology,2011,55(4-5):365-375.
[2]SAHARINEN P,BRY M,ALITALO K.How do angiopoietins Tie in with vascular endothelial growth factors?[J].Current Opinion in Hematology,2010,17(3):198-205.
[3]MAKINDE T,AGRAWAL D K.Intra and extravascular transmembrane signalling of angiopoietin-1-Tie2 receptor in health and disease[J].Journal of Cellular and Molecular Medicine,2008,12(3):810-828.
[4]WERMUTH C G.The Practice of Medicinal Chemistry[M].New York:Academic Press,2008: 549-571.
[5]刘靖,王林,杨晓明.多靶点蛋白酪氨酸激酶抑制剂的研究进展[J].国际药学研究杂志,2009,36(3): 161-171.
[6]HUDKINS R L,ZULLI A L,UNDERINER T L,et al.8-THP-DHI analogs as potent Type I dual TIE-2/VEGF-R2 receptor tyrosine kinase inhibitors [J].Bioorganic & Medicinal Chemistry Letters,2010,20(11):3356-3360.
[7]TIAN Y,XU J,LI Z,et al.Combined 3D-QSAR and docking modelling study on indolocarbazole series compounds as tie-2 inhibitors[J].International Journal of Molecular Sciences,2011,12(8):5080-5097.
[8]DANDU R,ZULLI A L,BACON E R,et al.Design and synthesis of dihydroindazolo[5,4-a] pyrrolo[3,4-c]carbazole oximes as potent dual inhibitors of TIE-2 and VEGF-R2 receptor tyrosine kinases[J].Bioorganic & Medicinal Chemistry Letters,2008,18(6):1916-1921.
[9]BECKNELL N C,ZULLI A L,ANGELES T S,et al.Novel C-3 N-urea,amide,and carbamate dihydroindazolo[5,4-a]pyrrolo[3,4-c]carbazole analogs as potent TIE-2 and VEGF-R2 dual inhibitors[J].Bioorganic & Medicinal Chemistry Letters,2006,16(20):5368-5372.
[10]UNDERINER T L,RUGGERI B,AIMONE L,et al.TIE-2/VEGF-R2 SAR and in vitro activity of C3-acyl dihydroindazolo[5,4-a]pyrrolo[3,4-c]carbazole analogs[J].Bioorganic & Medicinal Chemistry Letters,2008,18(7):2368-2372.
[11]SHEWCHUK L M,HASSELL A M,ELLIS B,et al.Structure of the Tie2 RTK domain: self-inhibition by the nucleotide binding loop,activation loop,and C-terminal tail [J].Structure,2000,8(11):1105-1113.
[12]JAIN A N.Surflex-Dock 2.1: Robust performance from ligand energetic modeling,ring flexibility,and knowledge-based search[J].Journal of Computer-Aided Molecular Design,2007,21(5):281-306.
[13]段爱霞,陈晶,刘宏德,等.分子对接方法的应用与发展[J].分析科学学报,2009,25(4): 473-477.
[14]唐建生.激酶抑制剂的计算机辅助设计[J].化学通报,2007,70(6): 471-475.
[15]SUN J,CAI S,YAN N,et al.Docking and 3D-QSAR studies of influenza neuraminidase inhibitors using three-dimensional holographic vector of atomic interaction field analysis[J].European Journal of Medicinal Chemistry,2010,45(3):1008-1014.
[16]JAIN A N.Surflex:fully automatic flexible molecular docking using a molecular similarity-based search engine[J].Journal of Medicinal Chemistry,2003,46(4):499-511.
[17]陈军,盛春泉,郑灿辉,等.VEGFR2活性腔性质以及与抑制剂的结合模式研究[J].化学学报,2007,65(6):547-552.
[18]伍小云,张嘉杰,吴曙光.VEGFR-2酪氨酸激酶抑制剂的三维定量构效关系研究[J].中山大学学报:自然科学版,2010,49(1):57-61.
猜你喜欢
同位素(2022年6期)2022-12-30
物理学报(2022年10期)2022-06-04
波谱学杂志(2021年3期)2021-09-07
山东农业大学学报(自然科学版)(2021年3期)2021-07-29
——以高中化学“氢键”的教学为例
教学月刊(中学版)(2020年13期)2020-12-29
唐山师范学院学报(2020年6期)2020-04-16
杭州化工(2020年2期)2020-01-16
世界农药(2019年2期)2019-07-13
唐山师范学院学报(2019年3期)2019-06-18
水运管理(2018年5期)2018-08-20
