
拓扑异构酶Ⅰ抑制剂的结构精析
2012-04-28 13:03朱海梅
首都医科大学学报 2012年3期
朱海梅 罗 勇 赵 明*
(1.首都医科大学化学生物学与药学院多肽及小分子药物北京市重点实验室,北京 100069;2.首都医科大学附属安贞医院泌尿科,北京 100029)
1 Introduction
The action mechanism of drugs has been an interesting issue of people worked in pharmacy,chemistry,biology and medicine.Exploring the binding mode and interactions between drugs and receptors is one of the ways to discover the mechanism of action of drugs.There are two major ways to get insight into the binding mode and interactions between the drug and its receptor.One is to resolve the X-ray co-crystal structures or NMR solution structures of the drug and its receptor,the other is to construct the complex structure of the drug and its recep-tor via molecular modeling techniques.With the assistance of molecular graphic softwares,the binding mode and interactions between the drug and its receptor can be visually observed at molecular level,which provides useful information in exposing the action mechanism of the drug and designing new drugs.Therefore,it is important for undergraduates, postgraduates, teachers and researchers in pharmacy,chemistry,biology and medicine to get the essential knowledge and master the relevant techniques and skills,such as follow the research progress,understand the relevant literature,collect useful structural information of bio-macromolecules from the library,skillfully operate molecular modeling and graphic software,and correctly analyze the mechanism of action of drugs.Here,taken nuclear DNA topoisomerases I as an example,we show that introducing the knowledge and techniques is an effective way to improve the level of education in universities.
Nuclear DNA topoisomerases are essentially involved in almost all vitally important cell processes,as well as in the chromosome compaction and segregation[1-3].They are responsible for the relaxation of the DNA superhelical tension that arises in cells as a result of several nuclear processes,including transcription,replication and recombination[1,3].It is well known that inhibiting the activity of topoisomerases is lethal and leads to cell death[3].The expression level and activity of human topoisomerase I(human Topo I)are usually higher in proliferating tumor cells than in normal cells.Therefore,it has been appointed by US National Cancer Institute(NCI)as one of the promising targets for anticancer drugs.
This paper reviewed the published data for human Topo I,which concerned the structure and mechanism of human Topo I,recent advances of its inhibitors and structural insights into the interfacial inhibition of human Topo I for the first time.
2 Action mechanism of human Topo I
Human Topo I relaxes DNA supercoils by a“controlled rotation mechanism”[1].A “controlled rotation mechanism”of human Topo I consists of three main steps.Firstly,human Topo I cleaves a single strand of DNA via a transesterification reaction.In this reaction the catalytic residue Tyr723 attacks a phosphodiester bond on a single strand of DNA[3-7],linking a 3'-phosphotyrosine with one side of the break while releasing a free hydroxylated strand[2].Thus,this reaction forms a covalent enzyme-DNA cleavable complex as the sole product.Secondly,human Topo I relaxes DNA by letting the 5'-OH end swivel around the intact strand and removes DNA supercoils.Then the broken DNA strand is resealed via another transesterification reaction.This reaction involves nucleophilic attack of the 3'-phosphotyrosine bond by the 5'-OH end of the free strand to restore a phosphodiester bond[3-7].Ultimately,human Topo I is released for the next catalytic cycle.As the energy of the phosphodiester bond is conserved in the covalent enzyme-DNA intermediate,the cleavage-religation step does not require ATP or divalent metal binding.
3 Inhibitors of human Topo I
Here,the mechanism of action of the mentioned inhibitors is either stabilizing Topo I-DNA cleavage complex and/or catalytically inhibiting the enzyme.
3.1 Camptothecin and derivatives
Camptothecin(CPT)is a pentacyclic alkaloid first isolated from the Chinese tree,Camptotheca acuminata[3].It specifically targets human Topo I by intercalating inside the Topo I-DNA cleavable complex[8],slowing the religation step and increasing the lifetime of the cleavable complex.Accumulation of cleavable complex increases the collision probability of the replication fork with the cleaved DNA strand,which in turn leads to a permanent double-strand DNA break and ultimately leads to programmed cell death[3].
Two water-soluble camptothecin derivatives,topotecan and irinotecan,have been used clinically for cancer treatment[3,8].Topotecan is prescribed for treating chemoresistant ovarian and small cell lung cancers[3,8-10].Irinotecan,a carboxylesterase-dependent prodrug,is administered with 5-fluorouracil and leucovorin for metastatic colorectal carcinomas.Its bispiperidine group has to be hydrolyzed to yield the active metabolite SN-38,which shows a clinical efficacy in the treatment of color-ectal cancer[3,8-10].
Camptothecins are the only clinically approved inhibitors of Topo Ⅰ[10].However,they have a series of limitations[3],such as they have relatively low water solubility;after drug removal,the Topo I-DNA-camptothecin cleavable complex is reversible and requires a long infusion time;camptothecins can be cross-resisted by the drug efflux membrane transporters ABCG2 and ABCB1 overexpressed by cells;in physiological conditions,camptothecins are chemically unstable since the a-hydroxy d-lactone ring of camptothecin can be hydrolyzed to carboxylate[10];the inactive carboxylate form of camptothecin is ready to bind to human serum albumin and decreases its availability for cellular uptake;and the watersoluble sodium salt of camptothecin needs to be cleared by kidneys,which causes hemorrhagic cystitis and myelotoxicity.The side effects(diarrhea and neutropenia)of camptothecins are dose-limiting and potentially severe.The limitations of camptothecins encouraged developing non-camptothecin inhibitors of human Topo I.
3.2 Non-camptothecin derivatives
More recently,the search[8]for non-camptothecin derivatives has led to developing various inhibitors of human Topo I,including the derivatives of indolocarbazole,indenoisoquinoline and phenanthridine. Compared to camptothecins,they are chemically more stable,lack the hydroxylactone moiety,and enable the formation of persistent human Topo I complexes[3].
Indenoisoquinolines are one of the three classes of non-camptothecin inhibitors of human Topo I in clinical development.Of indenoisoquinoline derivatives,NSC 725776 and 724998 are at clinical trials[10].
Phase I-Ⅱ clinical trials suggested that indolocarbazoles had additional cellular targets besides Topo I.The current clinical trials of them as anticancer drugs give no very positive results.
The phenanthridine derivatives have been licensed to Genzyme Inc.and will soon enter clinical trial of Phase I.Interestingly,ARC-111,the analog of phenanthridine,exhibits chemical and biological similarities with indenoisoquinolines[10].It will be interesting to compare the clinical activities of these two classes of noncamptothecin inhibitors of human Topo I.
4 Tertiary structure of human Topo I
Several X-ray crystal structures of human Topo I in complex with DNA and inhibitors have been determined[11-21].They advance our understanding of the structural arrangement and function of human Topo I,and provide insights into binding and inhibition patterns of several classes of inhibitors.
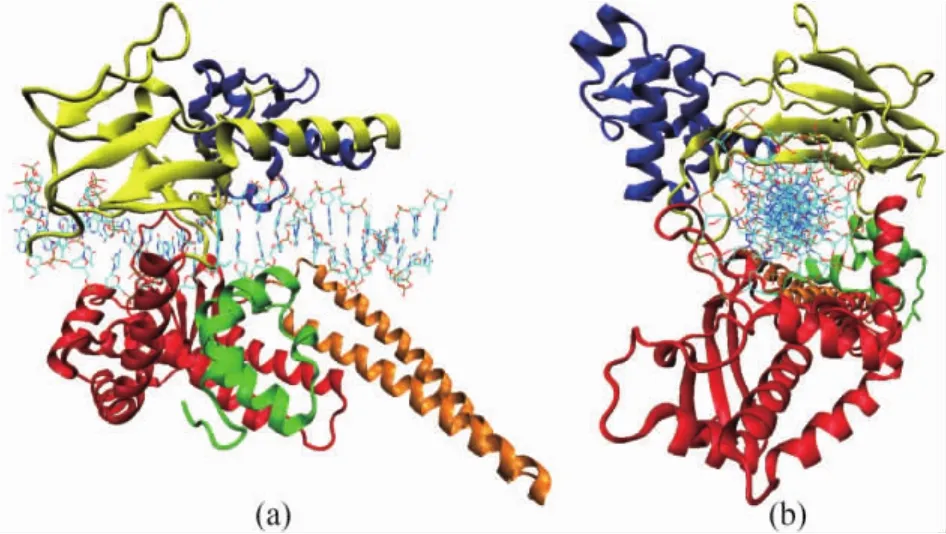
Fig.1 Tertiary structure of the human Topo I-DNA covalent complex viewed perpendicular to the pore(a)and down the pore(b).Core subdomains I,Ⅱ and Ⅲ are shown in yellow,blue and red ribbons respectively.Linker domain is shown in orange ribbons.The C-terminal domain is shown in green ribbon.DNA double strands are shown in bonds.The figure is created with PDB code 1K4T and rendered by VMD[22].
Human Topo I is a monomeric protein consists of 765 amino acid residues and contains four major regions(the N-terminal,core,linker and C-terminal domains)that clamp around the DNA.The core and the C-terminal domains are two highly conserved globes and are crucial for the catalytic activity.While the N-terminal and linker domains are not strictly essential for the catalytic and relaxation functions of human Topo I.The core domain combined with the C-terminal domain together are able to exhibit near full enzyme activity[11-13].The tertiary structure of the core,linker and C-terminal domains of human Topo I in covalent complex with DNA is shown in Fig.1.
The N-terminal domain consists of approximately 210 amino acid residues,is highly charged,is largely disordered,contains a few hydrophobic amino acids,Several nuclear-targeting signals are located in this domain.This domain has been implicated in nuclear localization through interactions with nucleolin[2].
The 56-kD core domain is composed of 425 amino acid residues(residues 210 to 635).This core domain contains subdomains I,Ⅱ,andⅢ (Fig.1).Subdomain I consists of residues 215 to 232 and 320 to 433,and is constructed by 2 helices and 9 strands.SubdomainⅡ,residues 233 to 319,is constructed by 5 helices and 2 strands[12].Subdomains I and Ⅱ are tightly together and bury about 1000 Å2of solvent-accessible surface area.SubdomainⅢ,residues 434 to 635,has a complex arrangement of 10 helices and 3 strands,and contains all the active-site residues,with the exception of the catalytic Tyr723[13].
The part of residues 636 to 712 is the linker domain that contains 2 long helices,contributes to but is not required for activity[12].
The C-terminal domain,residues 713 to 765,is constructed by 5 short helices,and contains the essential catalytic residue Tyr723.Tyr723 could form a phosphoester bond with the 3'phosphate at the cleavage site of the scissile strand of the substrate DNA[2,13].
The core domain of human Topo I interacts closely with its C-terminal domain.This largely hydrophobic interface buries about 1300 Å2of solvent accessible surface,is made up of 11 hydrophobic amino acid residues,5 specific hydrogen bonds and 3 salt bridges[11-12].
Human Topo I contains a central DNA-binding pore of 15 to 20 Å in diameter in its closed form.This central pore is composed of 15 lysines and 8 arginines,the positively charged residues,which give rise to a high positive electrostatic potential surrounding the pore[13].The catalytic residues,including Tyr723,are locked within this highly positively charged channel region.
The crystal structures show that via covalent and non-covalent bonds a central 10 bp of the 22 bp DNA oligonucleotides bind to the central pore of human Topo I,while the major contacts involve the DNA phosphate groups.Upon forming complex,the intimate interactions between the DNA oligonucleotides and human Topo I bury a total 4700 Å2of solvent accessible surface area(2400 Å2for human Topo I and 2300 Å2for the DNA)[11-13].Three subdomains(I,Ⅱ,andⅢ)of the core and the C-terminal domain interact with DNA,burying approximately 950,80,1050,and 330 Å2of solvent accessible surface area,respectively.In contrast to the quite limited interactions of DNA with subdomainⅡand the C-terminal domain,the interactions of DNA with subdomains I andⅢare very extensive[12-13].Subdomains I and Ⅲ are the large part of human Topo I's clamp that closes around the DNA before cleavage and covalent attachment.
5 Interfacial inhibition of human Topo I poisons
The X-ray crystal structures of the complexes of human Topo I with four kinds of inhibitors,including the camptothecins[13,20], indolocarbazoles[20], indenoisoquinolines[20], and norindenoisoquinolines[21], have been elucidated.These inhibitors,so called Topo I poisons,bind to a transient covalent complex of Topo I and DNA,and inhibit the resealing of a single-strand nick.Fig.2a shows the cleaved DNA double strand complexed with topotecan.Fig.2b is created with PDB codes 1K4T,1T8I,1T8L,1SC7 and 1SEU,and rendered by VMD[22].It shows the relative binding positions of camptothecin(c,yellow bonds),indenoisoquinoline(d,MJ238,green bonds),indolocarbazole(e,SA315F,orange bonds),and norindenoisoquinoline(f,purple bonds)to topotecan lactone(a,cyan bonds)and topotecan carboxylate(b,cyan bonds)in the crystal structures of Topo I-DNA-poison complexes.
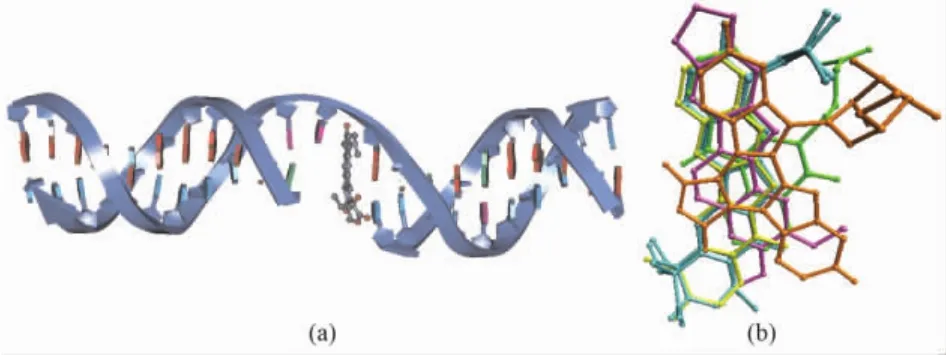
Fig.2 Binding mode of cleaved DNA double strand(navy ribbon)and topotecan(gray ball and stick)(a),and the superimposition of human Topo I poisons in the crystal structures of Topo I-DNA-poison complexes(b).The figure is rendered by DS2.5 and VMD[22].
Since the step of DNA religation is much faster than the step of DNA cleavage,in normal physiological condition,the covalent Topo I-DNA cleavage complex is barely detectable.On the other hand,several types of DNA alterations like single nucleotide gaps,basic sites,modi-fied bases and modified sugars can lead to persistent accumulation of the adducts of Topo I-DNA[3].
5.1 Binding mode of topotecan[3,8-10,13]
The structural features of camptothecins that are essential for activity include the 20(S)-hydroxyl,the pyridone moiety of the D-ring,the lactone moiety of the E-ring,and the planarity of the five-membered ring system.
Topotecan intercalates at the DNA cleavage site,forming base-stacking interactions with both the-1(upstream)and+1(downstream)base pairs.The planar system of the 5-membered polycyclic ring of topotecan mimics a DNA base pair of the DNA duplex.The 5-membered polycyclic ring occupies the same space as the+1 base pair in the structure without topotecan bound(a and b in Fig.3).About 60%of solvent accessible surface of topotecan(380 Å2of the total 630 Å2)is covered by base-stacking interactions.
There is only 1 direct hydrogen bond between topotecan and human Topo I,which is located between the hydroxyl of 20-C and the carboxylate oxygen of Asp533(a and b in Fig.3).Residue Asp533 forms a hydrogen bond to the immediate residue Arg364(a and b in Fig.3).Topotecan attaches to human Topo I in the minor groove region of the DNA.There are water-mediated hydrogen bonds between the hydrolyzed hydroxyacid form of topotecan and residues Asn722 and Tyr723(b in Fig.3).
5.2 Binding mode of camptothecin[3,10,20]
Camptothecin intercalates at the DNA cleavage site with a similar binding mode of topotecan.The E-ring of camptothecin is positioned closely toward the active site of human Topo I.The lactone oxygen of 21-C is 3.9 Å away from the bridging phosphodiester oxygen between Tyr723 and thymidine-10,and is 3.8 Å from the nitrogen of Lys532.The oxygen of pyridone ring is positioned 4.1 Å away from the side chain oxygen of Asn722.The hydroxyl of 20-C is positioned 3.2 Å from the side chain oxygen of Asp533.Residue Asp533 is required for enzyme sensitivity to camptothecin.The closest proteindrug interaction in the Topo I-DNA-camptothecin ternary complex is 2.9 Å,which is the hydrogen bond between the side chian nitrogen of Arg364 and a free electron pair of 1-N in the B-ring of camptothecin.Camptothecin forms two hydrogen bonds with human Topo I and intercalates between the DNA bases pairs(c in Fig.3).

Fig.3 Binding modes of(a)topotecan lactone form(cyan bonds),(b)carboxylate form(cyan bonds)and(c)camptothecin(yellow bonds)with human Topo I.The figure is rendered by VMD[22].
5.3 Binding mode of idenoisoquinolines[10,20]
MJ238 intercalates at the cleavage site of DNA,between the+1 and-1 base pairs.Rings A and B stack with the scissile strand bases,while rings C and D stack with the non-cleaved strand bases.The carbonyl of ring C is positioned on the minor groove side of the indenoisoquinoline ring and forms a bidentate interaction with the two nitrogens of the side chain of Arg364 at distances of 3.2 and 3.6 Å (d in Fig.4).The butyl-carboxylic acid substituent of the nitrogen of ring B is positioned in the major groove toward Asn352 and Ala351.
5.4 Binding mode of indolocarbazoles[3,8,20]
SA315F intercalates towards DNA at the cleavage site.SA315F is a symmetrical molecule with the exception of the linking of pyranosyl moiety to the nitrogen of indole.The glycosylated indole ring(IG)stacks with the bases of the intact strand side of the duplex DNA,which is similar to the stacking of rings A and B of camptothecin as well as rings C and D of MJ238.The nonglycosylated indole stacks with the bases of the cleaved strand side of the duplex DNA.The maleimide ring is located on the minor groove side of the intercalation binding pocket.One of the two carbonyls on the maleimide ring is 2.8 and 3.2 Å from two nitrogens of the side chain of Arg364,respectively(e in Fig.4).The hydroxyl of the glycosylated indole is positioned 3.8 Å from Glu356,whereas the hydroxyl of the nonglycosylated indole is positioned 3.6 Å from Ser722(e in Fig.4).A 3.0 Å distance between this hydroxyl and Asn722 would exist if the positon of Ser722 is replaced with Asn722.
5.5 Binding mode of noridenoisoquinolines[8,10,21]
The binding mode of norindenoisoquinoline to human Topo Ⅰ is similar to that of topotecan.Norindenoisoquinoline inserts at the cleavage site between the DNA base pairs of the ternary complex.The ternary complex is stabilized by stacking interactions and two direct hydrogen bonds to residues Arg364 and Asn722 of human Topo I(f in Fig.4).Norindenoisoquinoline also has a point of attachment to the enzyme in the minor groove region of the DNA similar to topotecan.

Fig.4 Binding modes of(d)indenoisoquinoline(green bonds),(e)indolocarbazole(orange bonds)and(f)norindenoisoquinoline(purple bonds)with human Topo I.The figure is rendered by VMD[22].
5.6 General principles of the binding mode of Topo I poisons[5-6,20,23]
The crystal structures of the ternary complexes of human Topo I with DNA and five classes of posions(topotecan,camptothecin,indolocarbazoles,indenoisoquinolines,and norindenoisoquinolines)suggest that the stacking interactions are crucial for efficient bonding of poisons to the covalent cleavable complex of Topo I and DNA.Some general principles of Topo I poison binding have been revealed and should be emphasized in the design of new posions of Topo I.The flat planar ring intercalate between the - 1/+1 base pairs at the Topo I-mediated DNA cleavage site.Only on the intact strand side of the duplex DNA,base stacking interactions are spatially conserved.On cleaved strand side of duplex DNA,variable rings and base-stackings are allowed.A free electron pair is important for hydrogen bonding on the minor groove side of the intercalation binding pocket near Arg364,even though the exact chemical property of the hydrogen bond acceptor could be very different.There are three distinct binding pockets,consisting of(a)a major groove in the vicinity of Asn352,(b)an intact strand side in the vicinity of Glu356,and(c)an“E-ring binding pocket”in the vicinity of the phosphotyrosine linkage to DNA and active site residues,for additional substituents.These results explain how the diverse chemotypes can adopt the same binding mode and exploit different features of the Topo I-DNA complex to poison Topo I.Therefore,it is possible to combine different features of each chemotype to develop novel poisons of Topo I.
[1]Forterre P,Gribaldo S,Gadelle D,et al.Origin and evolution of DNA topoisomerases[J].Biochimie,2007,89(4):427-446.
[2]Cyril V,Muller M T.A solid phase assay for topoisomerase I interfacial poisons and catalytic inhibitors[J].Anal Biochem,2012,421(2):607-616.
[3]Khadka D B,Cho WJ.3-Arylisoquinolines as novel topoisomerase I inhibitors[J].Bioorg Med Chem,2011,19(2):724-734.
[4]Bhat A G,Leelaram M N,Hegde S M,et al.Deciphering the distinct role for the metal coordination motif in the catalytic activity of mycobacterium smegmatis Topoisomerase I[J].J Mol Biol,2009,393(4):788-802.
[5]Nagarajan R,Kwon K,Nawrot B,et al.Catalytic phosphoryl interactions of topoisomerase IB [J].Biochemistry,2005,44(34):11476-11485.
[6]Khan Q A,Pilch D S.Topoisomerase I-mediated DNA cleavage induced by the minor groove-directed binding of bibenzimidazoles to a distal site [J].J Mol Biol,2007,365(3):561-569.
[7]Kennedy S,DiCesare J C,Sheaff R J.Topoisomerase I inactivation by a novel thiol reactive naphthoquinone[J].Biochem Biophy Res Comm,2011,410(1):152-158.
[8]Salerno S,Da Settimo F,Taliani S,et al.Recent advances in the development of dual topoisomerase I andⅡinhibitors as anticancer drugs[J].Curr Med Chem,2010,17(35):4270-4290.
[9]Teicher B A.Next generation topoisomerase I inhibitors:Rationale and biomarker strategies[J].Biochem Pharmacol,2008,75(6):1262-1271.
[10]Pommier Y.DNA topoisomerase I inhibitors:chemistry,biology,and interfacial inhibition [J].Chem Rev,2009,109(7):2894-2902.
[11]Hansen G,Harrenga A,Wieland B,et al.Crystal structure of full length Topoisomerase I from Thermotoga maritime[J].J Mol Biol,2006,358(5):1328-1340.
[12]González R D,Pertejo Y P,Redondo C M,et al.Structur-al insights on the small subunit of DNA topoisomerase I from the unicellular parasite Leishmania donovani[J].Biochimie,2007,89(12):1517-1527.
[13]Staker B L,Hjerrild K,Feese M D,et al.The mechanism of topoisomerase I poisoning by a camptothecin analog[J].PNAS USA,2002,99(24):15387-15392.
[14]Christmann-Franck S,Fermandjian S,Mirambeau G,et al.Molecular modelling studies on the interactions of human DNA topoisomerase IB with pyridoxal-compounds[J].Biochimie,2007,89(4):468-473.
[15]Pommier Y,Barcelo J M,Rao V A,et al.Repair of topoisomerase I-mediated DNA damage [J].Progress Nucleic Acid Res,2006,81:179-229.
[16]Punchihewa C,Dai J,Carver M,et al.Human topoisomerase I C-terminal domain fragment containing the active site tyrosine is a molten globule:Implication for the formation of competent productive complex [J].J Struct Biol,2007,159(1):111-121.
[17]Ganguly A,Sengupta S,BoseDasgupta S,et al.Mutational studies reveal lysine 352 on the large subunit is indispensable for catalytic activity of bi-subunit topoisomerase I from Leishmania donovani[J].Mol Biochem Parasit,2009,165(1):57-66.
[18]Interthal H,Quigley P M,Hol W G,et al.The role of lysine 532 in the catalytic mechanism of human topoisomerase I[J].J Boil Chem,2004,279(4):2984-2992.
[19]Chrencik J E,Staker B L,Burgin A B,et al.Mechanisms of camptothecin resistance by human topoisomerase I mutations[J].J Mol Biol,2004,339(4):773-784.
[20]Staker B L,Feese M D,Cushman M,et al.Structures of three classes of anticancer agents bound to the human topoisomerase I-DNA covalent complex [J].J Med Chem,2005,48(7):2336-2345.
[21]Ioanoviciu A,Antony S,Pommier Y,et al.Synthesis and mechanism of action studies of a series of norindenoisoquinoline topoisomerase I poisons reveal an inhibitor with a flipped orientation in the ternary DNA-enzyme-inhibitor complex as determined by X-ray crystallographic analysis[J].J Med Chem,2005,48(15):4803-4814.
[22]Humphrey W,Dalke A,Schulten K.VMD:visual molecular dynamics[J].J Mol Graph,1996,14(1):33-38.
[23]Chillemi G,D'Annessa I,Fiorani P,et al.Thr729 in human topoisomerase I modulates anti-cancer drug resistance by altering protein domain communications as suggested by molecular dynamics simulations [J].Nucleic Acids Res,2008,36(17):5645-5651.
猜你喜欢
高等理科教育(2022年2期)2022-05-09
中华养生保健(2020年10期)2021-01-18
祝您健康(2019年7期)2019-07-12
天然产物研究与开发(2018年9期)2018-10-08
中外医学研究(2016年32期)2017-02-27
中国实用医药(2016年29期)2016-12-26
医学研究杂志(2015年8期)2015-06-22
医学信息(2015年7期)2015-03-20
天然产物研究与开发(2012年4期)2012-12-22
食品工业科技(2012年10期)2012-10-24
