
不对称酰胺基液晶化合物的合成,表征及热分析
2012-04-01 01:57:34庄长福张加研吴春华秦永剑师同顺
化工技术与开发 2012年2期
庄长福 ,王 瑛 ,张加研 ,吴春华 ,秦永剑 ,师同顺
(1.西南林业大学材料工程学院,云南 昆明 650224;2.吉林大学化学学院,吉林 长春 130023)
卟啉化合物由于具有配位的多样性,优良的热稳定性以及多电子大共轭的分子结构等特征,而且易于合成,在材料化学、分子识别、药物化学、高选择性催化及仿生学等方面广泛应用,研究十分活跃[1~2]。卟啉液晶是一类功能型的盘状液晶,已受到研究者的高度重视。因为在柱状体系内的相邻共轭体系之间重迭较小导致电荷载流体的低流动性,卟啉液晶有望成为一种新型的有机光导体材料,有机半导体材料,非线性光学材料以及生物传感器,而且也是目前最好的自组装材料之一[3~4]。自Goodby在1980年第一次合成出卟啉液晶后,研究者对卟啉液晶的合成做了大量的工作,液晶性质也得到了广泛的研究。但目前为止,对于具有液晶性的中位取代四苯基卟啉及其衍生物,它们的侧链都是酰氧基、烷氧基、烷基等基团,而酰胺基侧链的卟啉液晶鲜有报导,且多为对称性卟啉液晶的文献报道,而对于含有肽键的不对称酰胺基卟啉液晶更是稀有。本文设计合成了一种含有肽键的新型不对称酰胺基卟啉液晶化合物,并通过紫外光谱、质谱、红外光谱、核磁共振氢谱、元素分析等现代分析方法表征了其结构,并研究了其热稳定性。
1 实验部分
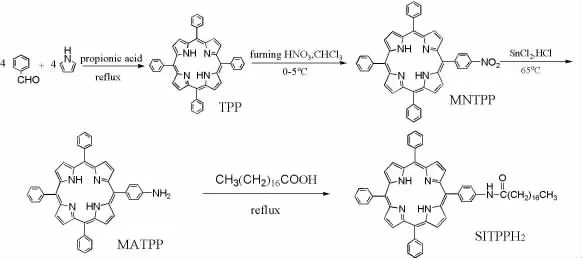
1.1 合成路线
SITPPH2的合成路线如下,5-(4-氨基)苯基-10,15,20-三苯基卟啉(MATPP) 按文献法[5]合成。
1.2 仪器和试剂
仪器:Varian Unity-500波谱仪、Nicolet 5PC FT-IR红外光谱仪(溴化钾压片)、Perkin-Elemer 240C自动元素分析仪、Shimadzu UV-240紫外-可见分光光度计、(TA SDT 2960)差热分析仪器(氮气气氛,加热速度 10℃·min-1,)、Axima CFRMALDITOF质谱仪。
试剂:所用化学试剂均是AR。
1.3 SITPPH2的合成
向三口瓶中加入15 mL二氯甲烷,然后溶解85 mg (0.30 mmol) 硬脂 酸 和 50 mg (0.41 mmol)4-二甲氨基吡啶,加3 mL三乙胺后冰浴搅拌,加入 80 mg (0.42 mmol)1-(3-二甲基氨基丙基)-3-乙基碳化二亚胺盐酸盐。混合均匀搅拌1 h后,向反应溶液中加入 95 mg (0.15 mmol) MATPP,常温反应12 h。加入150 mL蒸馏水,氯仿萃取,浓缩,硅胶柱层析, 乙酸乙酯和二氯甲烷(v∶v=1∶10)混合溶剂为淋洗液,目标产物为第一带紫色固体,收率 78%。1H NMR (300 MHz,CDCl3): 8.832~8.897 (m,8H,pyrrole ring),8.156~8.254 (m,8H,o-C6H4),7.872~7.943 (m,2H, m-phenyl),7.744~7.802 (m, 9H, p,m-triphenyl), 7.452 (s, 1H, -NH-CO-),2.503~2.575(m, 2H,-CO-CH2-), 1.813~1.921 (m, 2H, -CO-CH2-CH2-), 1.207~1.478(m,28H, alkyl CH2), 0.841~0.943(m,3H, alkyl CH3),-2.754 (s,2H,pyrrole N-H)。元素分析:实测值(计算值)/%:C 83.12 (83.09),H 7.43(7.31),N 7.78(7.81)。
2 结果和讨论
2.1 合成条件探讨
羧酸与氨基形成酰胺时要活化羧基,常见的方法有混和酸酐法,酰氯法,EDC或DCC法等。混合酸酐法优点是反应快方法简单且价格便宜,关键是反应条件的控制和选择,尤其是温度。酰氯法优点是反应较完全且速度快,但酰氯刺激性强毒性大,要求实验环境很严格。用EDC或DCC法反应条件温和,产率高且速率快。但DCC法后处理繁琐,杂质除净困难。而用EDC法生成水溶性副产物,水洗除去后柱层析即可。经过对比实验,用EDC为合成缩合剂。对于MATPP的合成主要有两条路线,一是通过混合醛法生成MNTPP,然后将MNTPP还原成MATPP;二是先生成TPP,再选择性硝化生成MNTPP,最后还原生成MATPP。实验对比发现虽然路线一比路线二步骤少,但路线一原料成本相对较高,且实验发现路线一制备出MNTPP产率低且分离繁琐,而路线二相比路线一虽然步骤多一步,但每步产率很高,容易控制反应过程,且原料相对便宜,后处理也相对容易,最终确定路线二为合成MATPP路线。
2.2 核磁共振氢谱和质谱
对卟啉化合物进行了核磁共振氢谱(1H NMR)的表征。图2是卟啉配体SITPPH2的核磁共振氢谱图,卟啉氢谱的明显特征是峰位置受卟啉环电流效应的影响而产生位移。卟啉配体SITPPH2的1H NMR 在 氘 代 氯 仿 测 得 ,δ-2.754 处 为 配 体SITPPH2卟啉环中吡咯N-H质子的特征峰,1.207~1.478 (m, 28H, alkyl CH2) 强峰的出现说明十八酸与氨基卟啉连接生成了SITPPH2,这是形成卟啉配体的标志之一。
图1是卟啉配体SITPPH2的质谱图,分子离子峰为 896.5,理论值为 896.21, 证明是目标化合物。
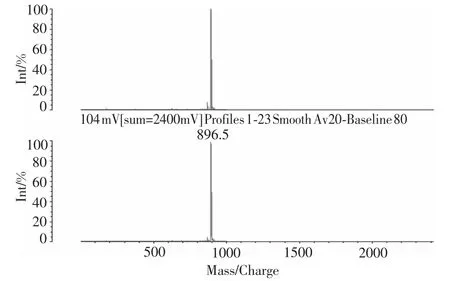
图1 SITPPH2的质谱图Fig.1 Mass spectra of SITPPH2
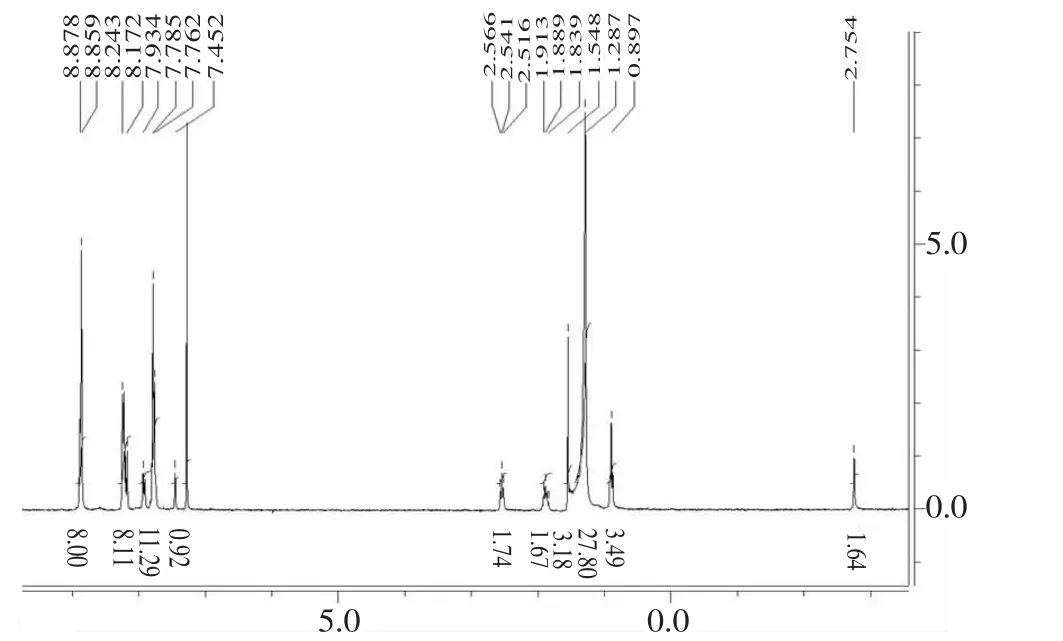
图2 SITPPH2的核磁共振氢谱图Fig.2 H NMR spectra of SITPPH2
2.3 紫外-可见光谱
紫外吸收光谱包括电子从基态跃迁至两个最低激发单重态Soret带和Q带,可见光区域(500~700 nm)产生4个弱Q带是由卟啉的a2μ(π)-e g(π*)跃迁产生的,为卟啉 π-π*跃迁的第一电子激发态;近紫外区(380~450 nm)产生强Soret带是由卟啉的 a1μ(π)-e g(π*)跃迁产生的,为卟啉 π-π*跃迁的第二电子激发态[6]。 在419 nm 是 SITPPH2的 Soret带,515、552、590、646 nm分别为4个Q带。而420 nm是MATPP的Soret带,515、555、590、650 nm 分别为 4 个 Q 带。从数据可以看出卟啉SITPPH2的Soret带和MATPP相比较没有明显差别。但是卟啉SITPPH2在551 nm和646 nm处的Q带和MATPP的相比发生了4nm的蓝移,说明卟啉SITPPH2的大环共轭程度没有明显变化。
2.4 红外光谱
图3是SITPPH2的红外光谱。SITPPH2的红外光谱在966 cm-1为卟啉环中心N-H的弯曲振动峰,3316 cm-1为伸缩振动峰,是卟啉化合物的特征红外吸收带。1666 cm-1是SITPPH2的酰胺Ⅰ带的羰基伸缩振动峰,1519 cm-1是酰胺Ⅱ带的振动峰,二谱带较宽可能是因为酰胺基与卟啉分子内的吡咯氢原子形成氢键所致[7]。因为在卟啉侧链上接有酰亚胺基团,酰亚胺的NH伸缩振动在3440cm-1和 3300 cm-1附近, 但由于卟啉配体SITPPH2中吡咯环位于3316 cm-1处的N-H伸缩振动比酰亚胺3300 cm-1的强,且二者波数相近,因此卟啉环的N-H伸缩振动将酰亚胺的N-H振动掩盖了[8]。卟啉吡咯环的C-N伸缩振动1350 cm-1对金属离子的取代非常敏感,当卟啉环嵌入金属离子后,C-N伸缩振动峰向低波数方向移动,是因为M-N的生成削弱了C-N键,使其力常数减小导致的[9]。MATPP氨基卟啉的 C-N振动峰1281 cm-1位移到 SITPPH2中的 1310 cm-1,是由于羧酸与MATPP的氨基形成了酰胺键,富电子基团氨基被吡嗪酰胺基离域致使电子平均化,使得C-N振 动 向 高 波 数 方 向 移 动[10]。 由 于 卟 啉SITPPH2的侧链中含有4个以上的亚甲基,因此在2920 cm-1和2850 cm-1处有亚甲基的反对称和对称伸缩振动强峰,723 cm-1处有亚甲基的面内摇摆振动峰。
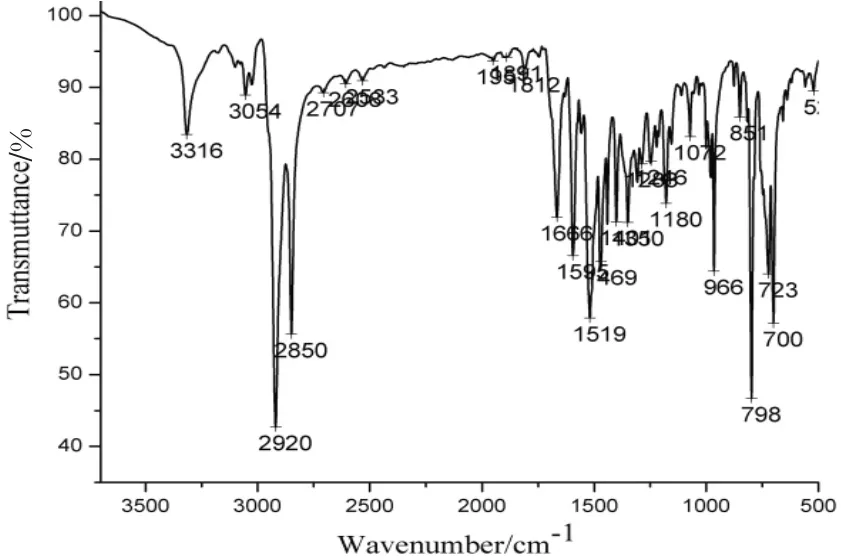
图3 SITPPH2的红外光谱Fig.3 IR spectra of SITPPH2
2.5 热分析
图4 是SITPPH2的热分析曲线。对于配体SITPPH2,小于200℃是稳定的,从200℃分解开始到340℃结束,失重了37.8%,对应的是失去卟啉侧链的十六酰氧苯基集团部分,非常接近理论值37.6%;而到 425℃时,失重为 64.5%,与理论值吻合,这个过程中DTA曲线在400℃有处吸热峰,对应失去十六酰氧苯基和3个苯基。卟啉化合物的分解是一个持续过程,DTA曲线在523℃有一处大吸热峰,425~550℃的区间包含了卟啉环和骨架的分解。在550℃以后,TGA曲线开始变得平滑。可以看出,卟啉SITPPH2有较好的热稳定性,在200℃下加热不分解,受热分解首先失去侧链部分,最后卟啉的骨架坍塌,是个持续过程。
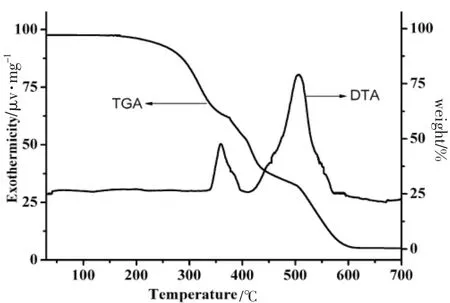
图4 SITPPH2的差热分析曲线Fig.4 TGA and DTA picture of SITPPH2
3 结论
不对称酰胺基液晶化合物卟啉5-(4-硬脂酸酰亚胺基)苯基-10,15,20-三苯基卟啉SITPPH2已被合成,其结构通过元素分析、质谱、紫外-可见光谱、核磁共振氢谱、红外光谱给予确认。热分析表明卟啉SITPPH2热稳定性很高,从200℃开始分解。
[1] Gust D,Moore T A, Moore A L,et al.Long-lived photoinitiated charge separation in carotene-diporphyrin triad molecules. [J].J Am Chem Soc, 991, 113:3638-3649.
[2] Johnson D G,Niemczyk M P,Minsek D W, et al.Photochemical electron transfer in chlorophyllporphyrin-quinone triads:the role of the porphyrinbridging molecule [J].J Am.Chem.Soc,1993,115:5692-5701.
[3] Li G Y, Che C, et al.Highly Selective Intra-and Intermolecular Coupling Reactions of Diazo Compounds to Form cis-Alkenes Using a Ruthenium Porphyrin Catalyst[J].Org Lett,2004, 6 (10): 1621-1623.
[4] Bonnett R.Photosensitizers of the porphyrin and phthalocyanine for photodynamic therapy.[J].Chem Soc Rew, 1995, 24:19-21.
[5] Wang D, Su L J, Cheng X L, et al.Synthesis and purification of porphyrin-schiff base with ethyl vanillin[J].Chemical Research in Chinese Universities, 2007,23 (2): 135-137.
[6] Ingar H.,Wasbotten, Jeanet C, et al.Electronic Absorption and resonance raman signatures of hyperporphyrins and nonplanar porphyrins [J] .J Phys Chem B, 2003, 107 (15): 3613-3623.
[7] Guo X.M.Synthesis and characterization of L-glutamic acid bridged porphyrin and its CD spectrum.[J].Chem J Chinese Universities, 2006, 27 (3): 410-413.
[8] Mo W M.The application of FT-IR in the synthesis of nicotinic derivative, [J].Chinese J. Spectroscopy Laboratory, 2004, 21 (4): 678-680.
[9] Fu S T.Synthesis and characterization of novel bridged porphyrin dimmers.[J].Chem.J Chinese Universities,2004,25 (7): 1204-1208.
[10] Jia H Y.Spectral study of 5, 10, 15, 20-tetra (pchlorophenyl)-porphyrin lanthanide complexes.[J].Chem.J Chinese Universities, 2004, 25 (2): 383-341.
猜你喜欢
云南化工(2023年7期)2023-08-01 07:59:34
现代临床医学(2022年4期)2022-09-29 07:36:10
中国抗生素杂志(2022年7期)2022-08-18 03:22:36
民用飞机设计与研究(2020年1期)2020-05-21 07:24:52
农药科学与管理(2019年8期)2019-11-23 08:04:44
长春师范大学学报(2019年4期)2019-04-29 05:51:36
中国塑料(2017年2期)2017-05-17 06:13:21
中国塑料(2015年6期)2015-11-13 03:02:49
化学工业与工程(2015年1期)2015-02-10 03:01:33
郑州大学学报(理学版)(2014年4期)2014-03-01 04:21:21
