
红霉素大环内酯合成通路在大肠杆菌中的重建
2012-02-09 09:34何彰华王洋叶丙雨师明磊王东范秋声黄芬赵志虎
生物工程学报 2012年2期
何彰华,王洋,叶丙雨,师明磊,王东,范秋声,黄芬,赵志虎
军事医学科学院生物工程研究所,北京100071
天然小分子药物一直是人类抵御疾病的重要手段,抗生素的发现及其使用,拯救了成千上万人的生命,但是大量抗性致病菌如耐万古霉素、甲氧西林抗性等“超级细菌”的出现,极大地威胁着人类生命健康[1]。一些危险的致病菌正对现有抗生素产生耐药性,它们进化的速度甚至超过了科学家开发新型药物的速度。因此如何建立组合性生物合成平台,更加有效利用现有天然小分子资源,开发新型抗生素,意义重大。
聚酮是来源于放线菌等的一大类结构复杂、活性多样、临床应用广泛的化合物。负责聚酮合成的模块性聚酮合成酶 (Polyketide synthase,PKS) 的多个基因成簇排列、活性位点的排列顺序与聚酮分子的合成步骤、产物结构间具有一一对应关系,特别适合利用基因工程等手段,进行组合性改造[2-3]。红霉素是一种典型的模块类聚酮抗生素,对红霉素结构的改造和修饰最有可能产生新型抗生素[4-6]。6-脱氧-红霉内酯B (6dEB)是红霉素生物合成过程中可分离出来的第一个中间体,其合成的分子机制已经研究得比较透彻,合成过程由红霉素聚酮合成酶模块介导完成[7],但在大肠杆菌中异源合成6dEB却存在几个难点[8]。2001年Pfeifer等[9]选择基因组中整合有枯草杆菌磷酸泛酰巯基转移酶基因sfp的大肠杆菌BAP1作为表达宿主,以实现聚酮合成酶中酰基运载蛋白 ACP的翻译后修饰;再应用天蓝色链霉菌的丙酰-CoA羧化酶基因 accA1、pccB以保障6dEB合成前体的供给;将合成6dEB必需的基因克隆于双质粒系统 pBP130和 pBP144 (表 3),首次在大肠杆菌 BAP1中以丙酸钠为底物异源合成了6dEB。
本研究旨在建立一个红霉素组合性生物合成的平台,即在大肠杆菌中重建红霉素大环内酯(6-脱氧-红霉内酯 B,6dEB) 合成通路。实验中先将参与6dEB合成所必需的基因分别克隆于多基因串联共表达载体[10],获得单基因重组质粒;再利用载体中 XbaⅠ/SpeⅠ互为同尾酶的特性实现相关基因的串联组合,获得多基因重组质粒pBJ130和 pBJ144。将多基因重组质粒共转化BAP1,获得含6dEB合成通路的工程菌株BAP1 (pBJ130/pBJ144),并对其进行了鉴定。研究中克服了高GC、长片段基因的PCR扩增,成功获取聚酮合成酶基因eryAI、eryAII、eryAIII;构建的多基因重组质粒中各基因分别位于独立的表达盒中,每个基因含单独的启动子和终止子,SDS-PAGE检测结果显示通路中各基因均有明显的表达;初步进行低温发酵,乙酸乙酯萃取后制样质谱检测到 6dEB,用红霉素标准品作内参得出其产量约 10 mg/L。研究成功实现了 6dEB合成通路在大肠杆菌中的重建,为红霉素大环内酯的改造和修饰提供了平台,也为红霉素合成通路在大肠杆菌中的完整重建以及聚酮类抗生素的组合性生物合成奠定了基础。
1 材料与方法
1.1 菌株和质粒
红霉素链霉菌Streptomyces erythraea购于中国农业菌种保藏管理中心;天蓝色链霉菌Streptomyces coelicolor购于 ATCC;表达宿主BAP1由塔夫茨大学化学与生物工程系 Blaine Pfeifer教授惠赠;大肠杆菌DH5a感受态和BL21感受态均购于天根公司;多基因串联表达载体pET-m22b (+) 和 pET-m28a (+) 由本实验室构建保存,(+) 代表含有f1复制区。
1.2 工具酶与化学试剂
T4 DNA连接酶购于NEB;DNA marker购于北京全式金生物公司;蛋白 marker为Fermentas产品;限制性内切酶和 Primer STAR聚合酶为TaKaRa产品;普通质粒小提试剂盒、琼脂糖凝胶回收试剂盒、普通DNA产物纯化试剂盒、细菌基因组DNA提取试剂盒均购于天根公司。
氨苄青霉素、卡那霉素购自AMRESCO公司;其他试剂均为国产分析纯产品。
1.3 研究中使用的基因
基因的信息均源自NCBI数据库。分子伴侣基因 groES-EL为本实验室克隆保存,其他基因为本实验克隆 (表1)。
1.4 引物设计与片段的扩增
引物的设计依照NCBI数据库中报道的基因序列,上、下游引物中分别设计合适的酶切位点用于克隆,具体信息见表2。
研究中的基因序列均富含 GC,且聚酮合成酶3个基因片段较长,约10 kb,PCR反应条件要求较严格。高GC、长片段PCR反应程序为 (两步法):94 ℃ 2 min;98 ℃ 10 s,70 ℃ 11 min,25个循环;72 ℃ 10 min,体系中加入5%的DMSO;高GC、短片段PCR反应程序为:94 ℃2 min;98 ℃ 10 s,64 ℃ 5 s,72 ℃ 90 s,30个循环;72 ℃ 5 min。
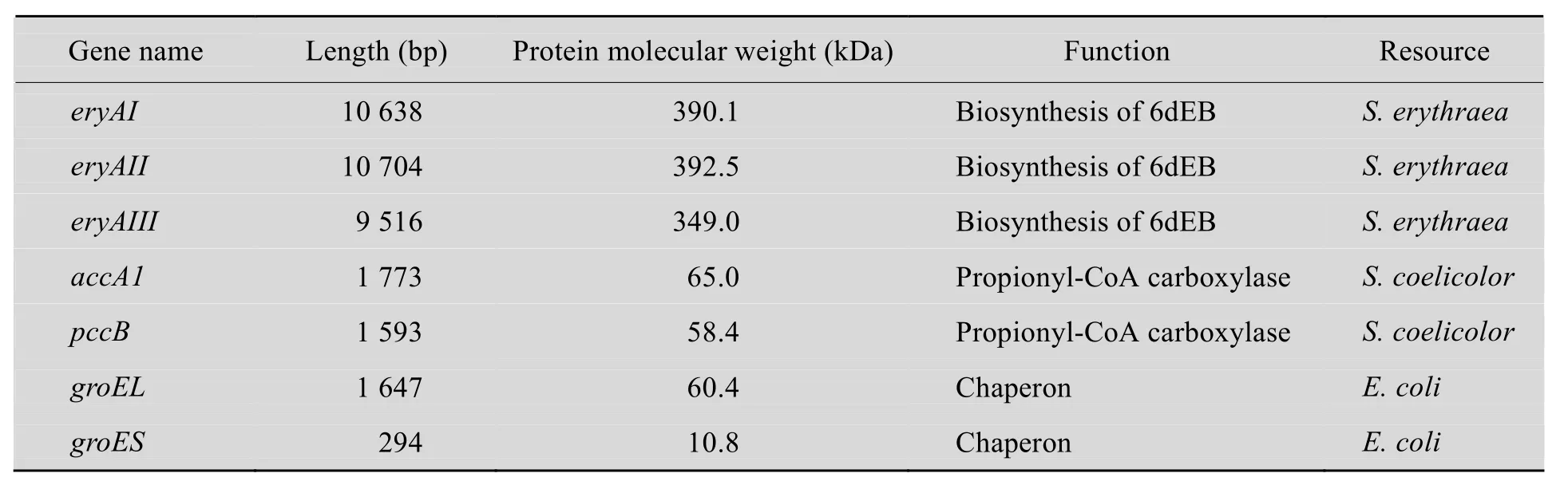
表1 文中所使用的基因Table 1 Gene used in the study

表2 文中所用引物及其序列Table 2 Primer and sequence used in the study
1.5 菌种的复苏培养与DNA的提取
菌种的复苏培养参照中国微生物菌种保藏管理委员会普通微生物中心提供的真空冷冻干燥菌种的恢复培养方法。复苏好的菌种挑单菌落转接入新鲜液体培养基,30 ℃振荡培养3~5 d,离心收集菌体,用细菌基因组DNA提取试剂盒提取全基因组 DNA。菌种的复苏培养采用高氏一号培养基。
1.6 重组质粒的构建
先将PCR获取的目的基因分别克隆入多基因串联表达载体pET-m28a (+) 或pET-m22b (+),进行酶切及测序鉴定;再利用多基因串联表达载体中XbaⅠ和SpeⅠ互为同尾酶的特性,通过酶切、连接实现相关基因的串联组合。多基因的组合策略如图1所示,组合模式见表3。
1.7 基因的表达鉴定
将构建成功的单基因重组质粒分别转化BL21进行诱导表达鉴定;将构建正确的多基因重组质粒pBJ130和pBJ144分别转化BAP1,用相应的抗生素筛选单克隆,获得表达菌株BAP1 (pBJ130) 和BAP1 (pBJ144);将重组质粒pBJ130和pBJ144共转化BAP1,用氨苄青霉素和卡那霉素筛选单克隆,获得含6dEB合成通路的工程菌株BAP1 (pBJ130/pBJ144)。诱导前先接菌至相应抗生素培养基中培养至饱和,诱导时按1%的比例转接至2 mL新鲜的培养基中,37 ℃培养约1 h,待OD600值达到0.4~0.6,加入IPTG (终浓度为0.5 mmol/L),于37 ℃继续培养4 h。各取1 mL菌液高速离心 1 min,沉淀悬于 100 μL 1×SDS凝胶上样缓冲液,沸水浴6 min,离心收集上清,取少量样品进行SDS-PAGE鉴定。
1.8 6dEB的合成与检测
接菌BAP1 (pBJ130/pBJ144) 至2 mL新鲜的氨苄青霉素和卡那霉素 LB培养基中37 ℃过夜培养。将活化的菌按 1%的接菌量转接至2 mL新鲜的LB (加双抗) 培养基中,37 ℃、250 r/min振荡培养,待OD600值达到0.6时将菌液于 22 ℃放置 5 min,加入 0.5 mmol/L IPTG、2.5 g/L丙酸钠,置于22 ℃、200 r/min继续培养72 h。
将上述低温诱导产物转移至离心管中12 000 r/min离心1 min,上清移至另一无菌的离心管中,加入等体积的乙酸乙酯萃取。将萃取的有机相在室温下风干,再溶于100 μL甲醇,样品送至军事医学科学院仪器中心进行质谱检测。在用乙酸乙酯萃取前,往发酵液中加入10 mg/L的红霉素标准品作内参[11]。
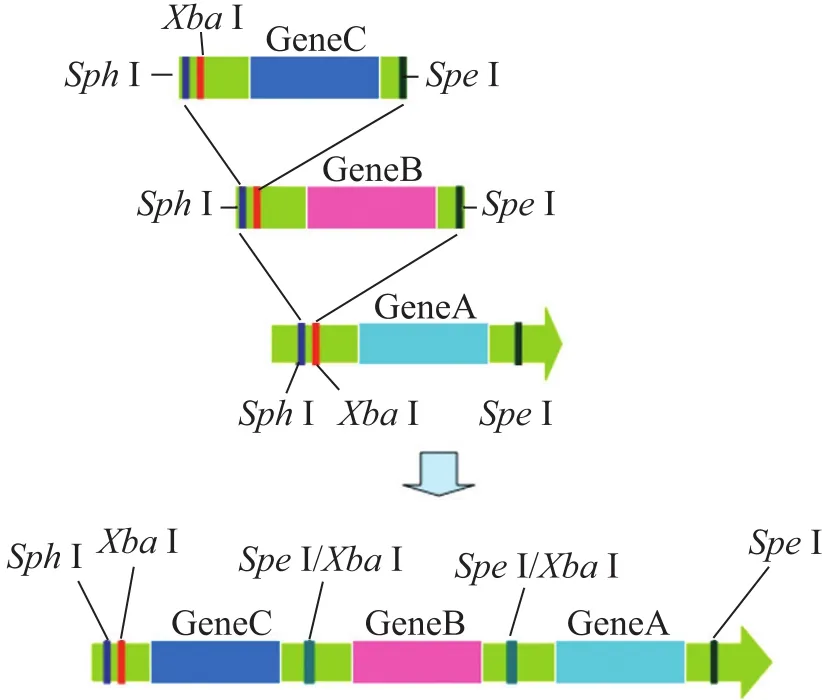
图1 多基因表达盒的串联策略Fig. 1 Schematic diagram of combination strategies.

表3 文中所用菌株和质粒Table 3 Strain and plasmid in the study
2 结果
2.1 目的基因的获取
以提取的红霉素链霉菌基因组 DNA为模板,用设计的引物选择 Primer STAR分别进行PCR扩增,成功获取 eryAI、eryAII、eryAIII三个基因,特异性均较好,结果见图2A~C。以提取的天蓝色链霉菌基因组DNA为模板,用设计的引物选用 Primer STAR聚合酶,成功扩取accA1和pccB,结果见图2D。
2.2 质粒的构建
2.2.1 构建单基因重组质粒
应用设计的酶切位点,将 PCR扩取的目的基因eryAI、accA1、pccB分别克隆入pET-m28a (+);eryAII、eryAIII分别克隆入pET-m22b (+)。用 Nde Ⅰ和 Hind Ⅲ分别对构建的单基因重组质粒进行双酶切鉴定,电泳结果见图3,均切下相应目的片段。利用通用引物并结合设计的测序引物将上述构建的单基因质粒分别进行测序鉴定,序列比对结果显示各基因均无碱基突变。
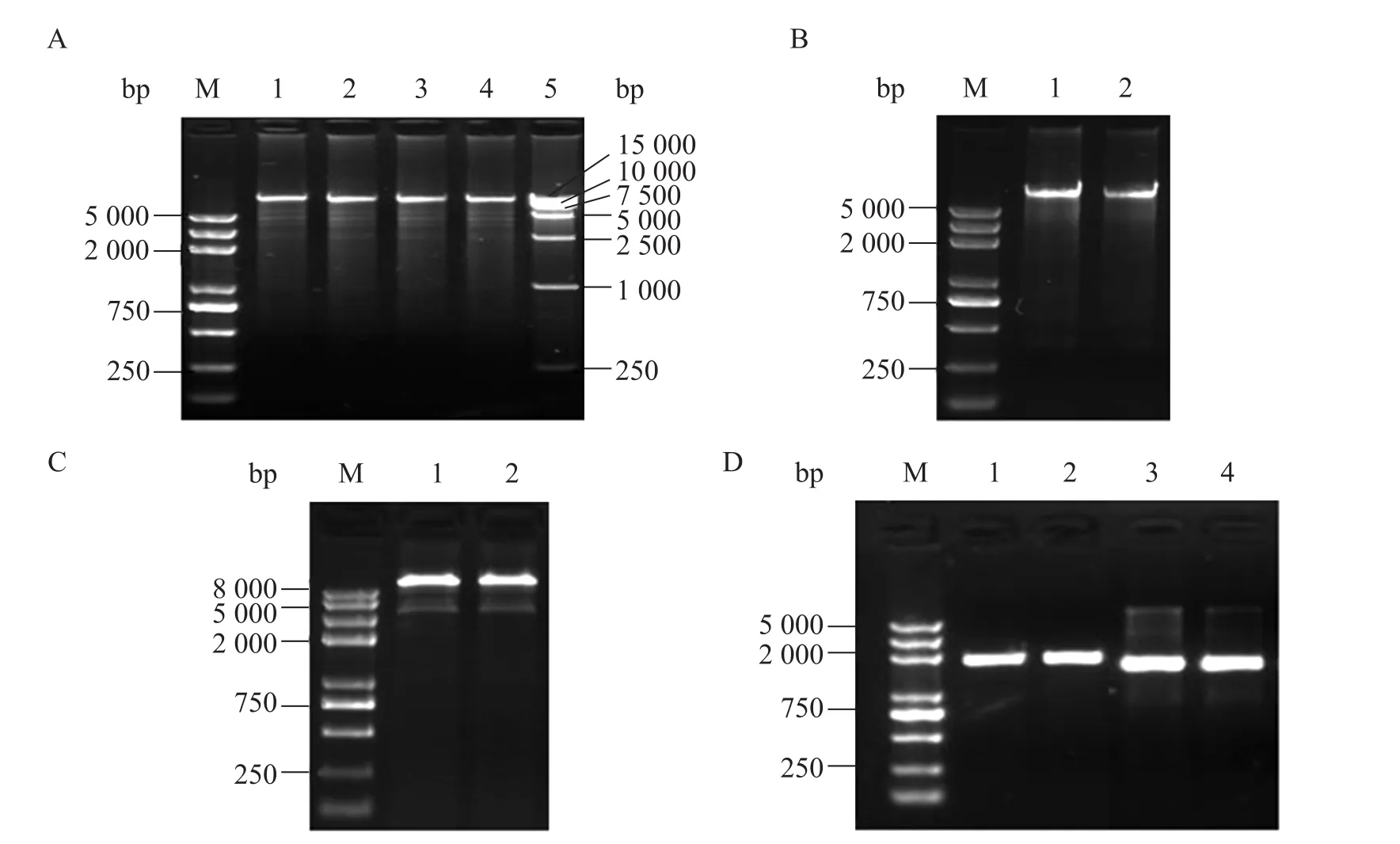
图2 目的片段的PCR扩增Fig. 2 PCR amplification of target fragment. (A) eryA. (B) eryAII. (C) eryAIII. (D) 1,2: accA1 PCR product; 3,4: pccB PCR product.
2.2.2 构建多基因质粒
利用XbaⅠ和SpeⅠ互为同尾酶的特性,选择合适的酶切位点 (如BglⅡ、Sph Ⅰ等),按图1所示多基因串联组合策略同时参照表3的组合模式完成相关基因的组合,获得多基因重组质粒pBJ130和pBJ144。用NdeⅠ分别酶切多基因重组质粒,电泳结果如图4所示,切下的基因片段均与预期一致。NdeⅠ单酶切后,基因片段上均带有约265 bp的连接序列。
2.3 基因的表达结果
2.3.1 单基因表达结果
单基因重组质粒转化大肠杆菌BL21后分别诱导,制备蛋白样品进行SDS-PAGE检测,目的蛋白条带清晰,大小正确,单个基因均能在大肠杆菌中表达 (图5)。
2.3.2 多基因共表达结果
诱导后,制备蛋白样品进行 SDS-PAGE检测,在大肠杆菌BAP1中相应的基因均有较明显的表达,其中 eryAI、eryAII、eryAIII分子量较大,表达量相对较低。编号2中eryAII、eryAIII条带均较明显,eryAII表达量相对较少;编号3中5个基因共表达,groEL、pccB分子量接近条带重叠,accA1条带均较明显,eryAI表达量相对较低,groES分子量较小已跑出胶底;编号 4中7个基因共表达,其中eryAI、eryAII分子量接近两蛋白条带重叠,groEL、pccB分子量接近两蛋白条带重叠,accA1、eryAIII条带均较明显,groES分子量较小已跑出胶底 (图6)。
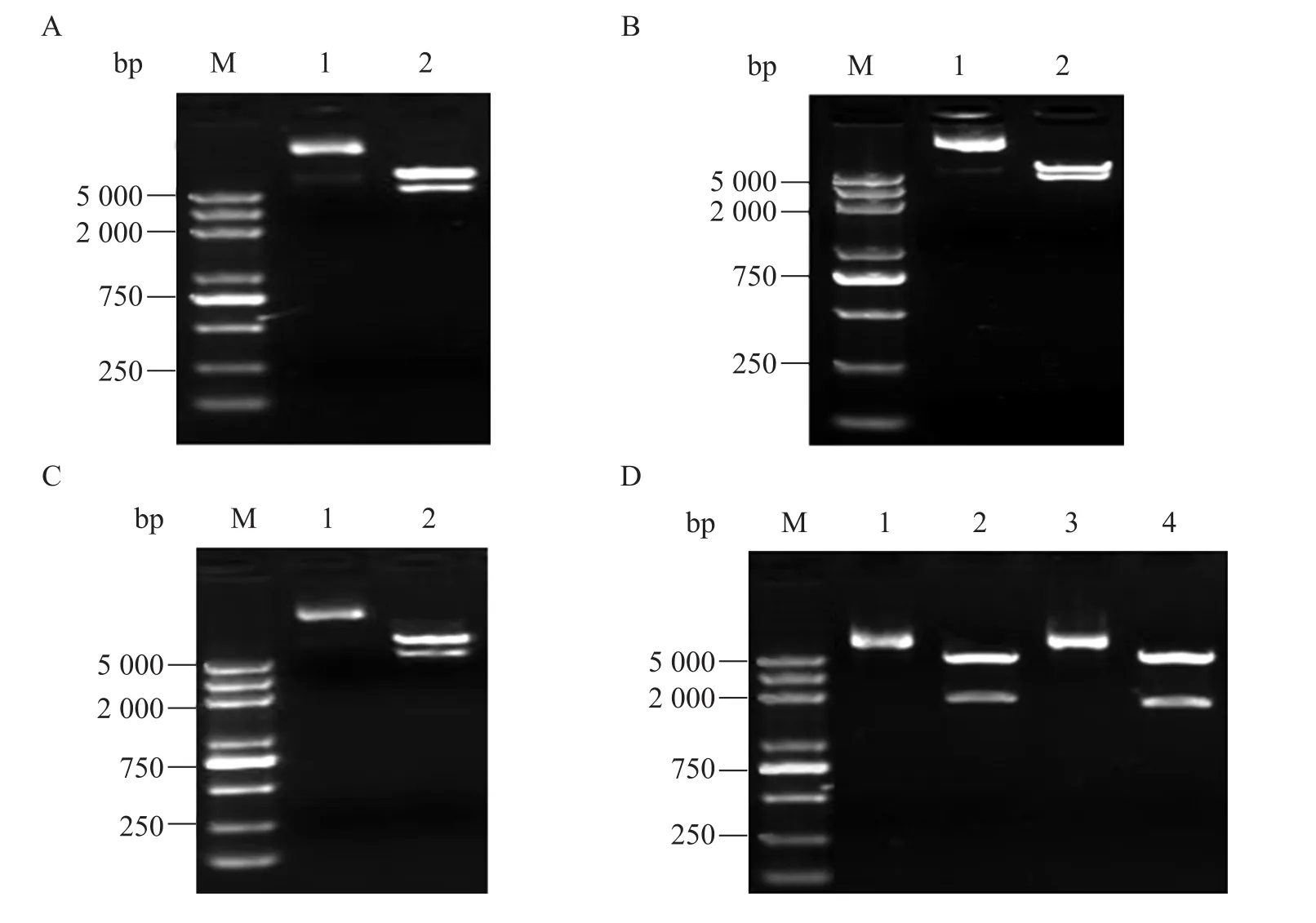
图3 单基因重组质粒的酶切鉴定Fig. 3 Restriction digestion analysis of recombinant plasmid. M: DNA marker. (A) 1: pET-m28a-eryAI; 2: pET-m28a-eryAI/Nde I+Hind III. (B) 1: pET-m22b-eryAII; 2: pET-m22b-eryAII/Nde I+Hind III. (C) 1: pET-m22b-eryAIII; 2: pET-m22b-eryAIII/Nde I+Hind III. (D) 1: pET-m28a-accA1; 2: pET-m28a-accA1/Nde I+Hind III; 3: pET-m28a-pccB; 4: pET-m28a-pccB/Nde I+Hind III.
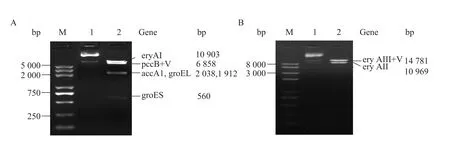
图4 多基因重组质粒的酶切鉴定Fig. 4 Enzyme digestion analysis of multi-genes recombinant plasmid. M: DNA marker. (A) 1: pBJ144; 2: pBJ144/Nde I. (B) 1: pBJ130; 2: pBJ130/Nde I. V shows vector.
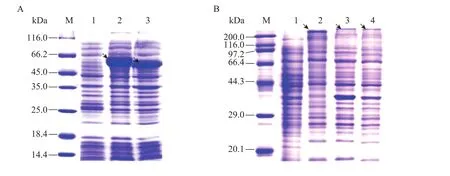
图5 SDS-PAGE检测单基因表达结果Fig. 5 SDS-PAGE analysis of the induced single genes expression individually. M: protein marker. (A) 1: BL21 (DE3)/pET-m28a-accA1 uninduced; 2: BL21 (DE3)/pET-m28a-accA1 induced; 3: BL21 (DE3)/pET-m28a-pccB induced. (B) 1: BL21 (DE3)/pET-m22b-eryAIII uninduced; 2: BL21 (DE3)/pET-m22b -eryAIII induced; 3: BL21 (DE3)/ pET-m22b-eryAII induced; 4: BL21 (DE3)/pET-m28a-eryAI induced. Arrow shows target protein.

图6 SDS-PAGE检测多基因共表达结果Fig. 6 SDS-PAGE analysis of the induced multi-genes expression simultaneously. M: protein marker; 1: BAP1 (pBJ130) uninduced; 2: (pBJ130) induced; 3: BAP1 (pBJ144) induced; 4: BAP1 (pBJ144/pBJ130) induced. Arrow shows interest protein.
2.4 6dEB的合成与检测
收集BAP1 (pBJ144/pBJ130) 低温诱导产物,萃取后风干再溶于甲醇,将样品送去做质谱检测。质谱结果如图 7所示:在 409.2565处出现6dEB的加钠峰,734.4753为10 mg/L红霉素标准品的质谱峰;6dEB的峰值略高于内参红霉素标准品,初步判断6dEB的产量约10 mg/L。

图7 质谱检测结果Fig. 7 Mass spectrum results of 6dEB. shows target profiles.
3 讨论
本研究应用多基因串联表达载体实现了参与 6dEB合成的多个基因在大肠杆菌中的共表达,完成了6dEB合成通路在大肠杆菌中的重建,首次在国内实现以丙酸钠为底物在大肠杆菌中异源合成了 6dEB。初次诱导发酵,以红霉素标准品作内参得其产量约 10 mg/L,这与 2001年Pfeifer等[9]获得的产量基本一致。研究借鉴了Pfeifer等[9]的表达策略,但在重组质粒的构建过程中我们做了适当的改善,应用了本实验室构建的多基因串联共表达载体,在本研究构建的6dEB合成通路中,各基因分别位于独立的表达盒中,每个基因含单独的启动子和终止子 (表3),避免了多基因共用启动子时的翻译极性效应,有望在后期的研究中通过调节启动子强弱对基因的表达进行更为精细的调控,以提高6dEB在大肠杆菌中的合成效率。
研究中的红霉素链霉菌及天蓝色链霉菌基因组GC含量比较高,而富含GC序列的DNA 片段特别是长片段的PCR扩增通常存在一定困难。在早期的研究中,我们选择Roche公司的Expand Long Template PCR System,通过不同条件的优化,扩增了5 kb富含GC的基因[12];使用Finzyme公司的DynaZyme EXT,在PCR体系中加入10%的DMSO,成功扩取了聚酮合成酶基因eryAIII,但特异性不理想[13]。本实验中,我们再次对高GC、长片段基因的PCR扩增进行了探索,使用TaKaRa公司的 Primer STAR聚合酶成功实现eryAI、eryAII、eryAIII的特异性扩增。将PCR获取的目的基因分别克隆入多基因串联共表达载体后,酶切鉴定无误,测序结果显示各基因均无碱基突变。
在大肠杆菌中构建6dEB合成通路,需要涉及多个基因的共表达,且巨大蛋白在大肠杆菌中的高效表达,是一个比较棘手的问题。研究中合理的应用多基因串联表达载体,便捷地实现了相关基因的串联组合;通过分子伴侣的共表达,可以促进外源蛋白的折叠,增加巨大蛋白的可溶性[14]。单基因的诱导表达鉴定结果显示:参与6dEB合成的相关基因均能在大肠杆菌中异源表达,且表达量较高 (图5)。在多基因的诱导共表达鉴定结果中,由于部分蛋白分子量接近而发生条带重叠现象,且分子量较大的基因 eryAI、eryAII、eryAIII表达量相对较低,但仔细对比各泳道中目的蛋白的表达情况可以初步判断各基因均能协同表达。发酵产物的质谱分析直接证实相关基因的确都正确表达了,同时这也进一步证实了本实验室构建的多基因串联共表达载体有着重要的应用前景,可以高效介导多基因、大片段基因的共表达。
6dEB在大肠杆菌中的成功合成意义重要,可以认为是红霉素基因工程研究的一个转折点。至 2001年 Pfeifer等[9]在大肠杆菌中异源合成6dEB以来,红霉素基因工程的研究有了迅猛的发展。在提高6dEB 合成产量方面,Lau等[15]采用高细胞密度分批发酵的方法,用优化的培养基使6dEB的产量上升至1.1 g/L;另有一些研究者在基因的表达调控方面进行了探索,均大幅度提高了 6dEB 在大肠杆菌中的合成产量[16-17]。在6dEB后修饰的研究中,Peiru等[11]和 Lee等[18]分别选取来源于其他链霉菌中的糖基化基因,构建了6dEB的后修饰通路,以6dEB 为底物在大肠杆菌中异源合成了Ery D和6dEry D等;2010年Pfeifer等[19]将红霉素链霉菌基因组中参与红霉素糖基化的18个基因克隆于3个不同抗性的质粒中,以6dEB为底物合成了Ery A。此外,Menzella等[20]合成了红霉素PKS基因序列,通过改造和替换PKS中的模块,合成了非天然的天然产物。
本研究完成了6dEB合成通路在大肠杆菌中的重建,建立了红霉素大环内酯改造和修饰的平台,也为之后红霉素合成通路在大肠杆菌中的完整重建以及聚酮类抗生素的组合性生物合成奠定了坚实的基础。
致谢:感谢军事医学科学院毒药物研究所王好山和张诚老师在质谱检测方面的帮助和指导;感谢军事医学科学院生物工程研究所吴军研究员和张惟材研究员为本实验提供了低温诱导条件!
REFERENCES
[1] Fischbach MA, Walsh CT. Antibiotics for emerging pathogens. Science, 2009, 325(5944): 1089−1093.
[2] Keaeing TA, Walsh CT. Initiation, elongation, and termination strategies in polyketide and polypeptide antibiotic biosynthesis. Curr Opin Microbiol, 1999, 3(9): 598−606.
[3] Wu N, Kudo F, Cane DE, et al. Analysis of the molecular recognition features of individual modules derived from the erythromycin polyketide synthase. Am Chem Soc, 2000, 122(20): 4847−4852.
[4] Kuhstoss S, Huber M, Turner JR, et al. Production of a novel polyketide through the construction of a hybrid polyketide synthase. Gene, 1996, 183(1-2): 231−236.
[5] Ruan X, Pereda A, Stassi DL, et al. Acyltransferase domain substitutions in erythromycin polyketide synthase yield novel erythromycin derivatives. J Bacteriol, 1997, 179(20): 6416−6425.
[6] McDaniel R, Thamchaipenet A, Gustafsson C, et al. Multiple genetic modifications of the erythromycin polyketide synthase to produce a library of novel“unnatural” natural products. Proc Natl Acad Sci USA, 1999, 96(5): 1846−1851.
[7] Zhang BC, Zhao ZH, Ma QJ. Molecular biology on erythromycin biosynthesis. Lett Biotech, 2001, 12(2): 151−160.张部昌, 赵志虎, 马清钧. 红霉素生物合成的分子生物学. 生物技术通讯, 2001, 12(2): 151−160.
[8] Zhang BC, Li LL, He BK, et al. Research advance in application of Escherichia coli expression system in erythromycin genetic engineering. Bull Acad Mil Med Sci, 2003, 27(5): 381−384.张部昌, 李凌凌, 贺秉坤, 等. 大肠杆菌表达系统在红霉素基因工程中应用的研究进展. 军事医学科学院院刊, 2003, 27(5): 381−384.
[9] Pfeifer BA, Admiraal SJ, Gramajo H, et al. Biosynthesis of complex polyketides in a metabolically engineered strain of E. coli. Science, 2001, 291(5509): 1790−1792.
[10] He ZH, Wang Y, Zhao J, et al. Construction of a vector suitable for the tandem coexpression of multiple genes by a single plasmid. China Biotechnol, 2011, 31(1): 40−45.何彰华, 王洋, 珺赵 , 等. 一种多基因串联共表达载体的构建. 中国生物工程杂志, 2011, 31(1): 40−45.
[11] Peirú S, Menzella HG, Rodríguez E, et al. Production of the Potent Antibacterial Polyketide Erythromycin C in Escherichia coli. Appl Environ Microbiol, 2005, 71(5): 2539−2547.
[12] Zhang BC, Zhao ZH, Yu XQ, et al. Optimizing conditions for PCR amplification of DNA with rich GC. Bull Acad Mil Med Sci, 2002, 26(4): 257−261.张部昌, 赵志虎, 于秀琴, 等. 富含GC DNA PCR扩增条件的优化. 军事医学科学院院刊, 2002, 26(4): 257−261.
[13] Zhang LH, Wang Y, He ZH, et al. Cloning and expression of polyketide synthases gene eryAIII of Saccharopolyspora erythraea in Escherichia coli. Lett Biotech, 2010, 21(6): 794−797.张丽华, 王洋, 何彰华, 等. 红霉素链霉菌聚酮合成酶基因 eryAIII的克隆及其在大肠杆菌中的表达. 生物技术通讯, 2010, 21(6): 794−797.
[14] Zhang JY, Zhao ZH, Cai MH. Progress in molecular chaperon GroEL. Lett Biotech, 2001, 12(2): 127−129.张经余, 赵志虎, 蔡民华. GroEL分子伴侣研究进展. 生物技术通讯, 2001, 12(2): 127−129.
[15] Lau J, Tran C, Licari P, et al. Development of a high cell-density fed-batch bioprocess for the heterologous production of 6-deoxyerythronolide B in Escherichia coli. J Biotechnol, 2004, 110(1): 95−103.
[16] Wang Y, Boghigian BA, Pfeifer BA. Improving heterologous polyketide production in Escherichia coli by overexpression of an S-adenosylmethionine synthetase gene. Appl Microbiol Biotechnol, 2007, 77(2): 367−373.
[17] Zhang HR, Boghigian BA, Pfeifer BA. Investigating the role of native propionyl-CoA and methylmalonyl-CoA metabolism on heterologous polyketide production in Escherichia coli. Biotechnol Bioeng, 2010, 105(3): 567−573.
[18] Lee HY, Khosla C. Bioassay-guided evolution of glycosylated macrolide antibiotics in Escherichia coli. PLoS Biol, 2007, 5(2):243−250.
[19] Zhang HR, Wang Y, Wu JQ, et al. Complete biosynthesis of erythromycin A and designed analogs using E. coli as a heterologous host. Chem Biol, 2010, 17(11): 1232−1240.
[20] Menzella HG, Reid R, Carney JR, et al. Combinatorial polyketide biosynthesis by de novo design and rearrangement of modular polyketide synthase genes. Nat Biotechnol, 2005, 23(9): 1171−1176.
猜你喜欢
保健与生活(2022年7期)2022-04-08
华声文萃(2021年11期)2021-11-15
昆明医科大学学报(2020年12期)2021-01-26
国际呼吸杂志(2019年4期)2019-03-12
新世纪智能(英语备考)(2018年11期)2018-12-29
山东医药(2017年20期)2017-07-01
中国环境监察(2016年7期)2016-10-23
中国现当代社会文化访谈录(2016年0期)2016-09-26
中国学术期刊文摘(2016年1期)2016-02-13
探测与控制学报(2015年4期)2015-12-15
