
有机/无机复合微胶囊相变材料的近红外光谱分析
2011-11-30 10:41吴晓琳朱朋莉杜如虚
物理化学学报 2011年5期
吴晓琳 孙 蓉 朱朋莉 杜如虚
(1中国科学院深圳先进技术研究院,广东深圳518055; 2中国科学院研究生院,北京100049; 3香港中文大学,香港)
有机/无机复合微胶囊相变材料的近红外光谱分析
吴晓琳1,2孙 蓉1,*朱朋莉1杜如虚3
(1中国科学院深圳先进技术研究院,广东深圳518055;2中国科学院研究生院,北京100049;3香港中文大学,香港)
通过傅里叶变换近红外(FT-NIR)光谱分析技术,探索聚甲基丙烯酸甲酯-二氧化硅@相变材料(PMMA-SiO2@PCM)微胶囊相变过程的光谱学特性和相变机理,分析相变过程微胶囊的微结构变化特性.结果显示:微胶囊中,石蜡的融化过程就是-CH2对称伸缩振动逐渐增强和非对称伸缩无规则振动共存的振动变化过程.石蜡相变过程中,其近红外吸收峰强度的变化仅是壳层材料吸收峰强度变化幅度的一半.同时,近红外光谱可以用来辅助分析微胶囊的核壳结构,实现微胶囊相变过程监测.近红外光谱在微胶囊相变材料相变过程的应用对相变机理的研究及高效相变材料的选择具有重要的科学意义和应用价值.
微胶囊;傅里叶变换近红外光谱;相变材料;相变;有机/无机复合材料
1 引言
傅里叶变换近红外(FT-NIR)光谱因具有分析速度快、产出多,可以用于样品的定性、定量分析;具有不破坏样品、不用试剂、无污染、穿透性高等特性,在果品质量检测、药物在线检测分析、石油化工、聚合物等领域得到了广泛应用.1与常用的中红外光谱相比较,近红外光谱可以实现大量样品的原位快速在线分析,具有更深的穿透性,能实时检测物质的微结构变化信息.可直接实现大量产品的过程分析,避免微量分析带来的产品质量检测误差.微胶囊相变材料(MEPCM)2-4是以相变材料(PCM)为芯材,由有机聚合物5-11或无机材料12-18为壳层的胶囊结构19-21组成的.其相变过程是连续的物理变化过程.常用的差示热扫描量热仪(DSC)只能宏观地观察到少量微胶囊相变材料相变温度及相变潜热情况,无法检测到相变过程微胶囊的微结构变化信息.同时,即使相变材料未能被成功地包覆,其DSC结果也是可以比较稳定的循环,无法辨别是否是核壳结构.近红外光谱可以补偿DSC分析手段的不足.它是从微观结构的角度分析大量微胶囊相变材料相变过程的微结构变化信息.而且,近红外光谱可以将核壳结构和非核壳结构通过分子结构吸收信号的变化来加以分辨.因此,将近红外光谱在线分析技术与DSC分析结果相结合,可以得到微胶囊相变过程较详细的重要的热物特性信息,包括微胶囊相变材料的相变特性、微结构变化特性、核壳结构特性等.而热物特性和微结构特性是微胶囊相变材料应用的主要参数.故,近红外光谱和DSC在微胶囊相变材料相变过程的应用对相变机理研究、核壳结构的分析及高效相变材料的选择具有重要的理论意义和应用价值.
本文采用了FT-NIR光谱技术与DSC分析结果相结合的分析方法,对微胶囊相变材料相变过程的结构变化进行原位的实时监测,探讨相变过程的微结构变化特性及相关机理.实验采用一步法,快速而高效地制备有机/无机复合材料.实验中,有机聚合物选择具有高冲击强度和较佳化学稳定性的聚甲基丙烯酸甲酯(PMMA).12无机物选择化学稳定性好且制备技术较为成熟的SiO2.本实验引用了既可以作为单体引发剂又可作为酯类水解催化剂的过硫酸铵,使单体的界面聚合反应和酯类的界面水解反应同时进行,从而提高有机/无机微胶囊的结构均匀性和稳定性.
2 实验部分
2.1 材 料
实验选择南阳石化有限公司的工业纯石蜡(PCM)为相变材料,天津光复精细化工研究所的分析纯甲基丙烯酸甲酯和天津大茂化学试剂厂的分析纯正硅酸乙酯为聚合反应和水解反应的前驱体,天津光复精细化工研究所的分析纯过硫酸铵为引发剂,天津大茂化学试剂厂的分析纯十二烷基苯磺酸钠为表面活性剂.
2.2 实验过程
2.2.1 PMMA-SiO2@PCM微胶囊的制备
将油相的石蜡、甲基丙烯酸甲酯(MMA)和正硅酸乙酯以6:1:9质量比混合,并用超声均匀分散2 min.油相混合溶液均匀分散后,添加到圆底烧瓶中,并加热至60°C,机械搅拌速率为1500 r·min-1.将配置好的1.5%(w)的十二烷基苯磺酸钠水溶液42.5 mL添加到圆底烧瓶中乳化10 min后,水浴温度调至85°C,再乳化20 min,至形成均匀的乳白色微乳液.最后,将浓度为0.13 g·mL-1,pH约为3的引发剂和催化剂过硫酸铵水溶液7.5 mL缓慢地向圆底烧瓶中滴加至完全.随后,将机械搅拌的速率调为1000 r·min-1,水浴的温度调至80°C.1.5 h后,水解反应和聚合反应进行完全.向降至室温的微胶囊乳液中添加饱和的NaCl水溶液10 mL,并静置30 min后,会有分层现象.把分层的乳液通过真空抽虑的方式用蒸馏水洗涤3次,并用真空干燥箱在45-50°C的条件下真空干燥4 h,最终得到白色的PMMASiO2@PCM微胶囊粉体.
2.2.2 PMMA-SiO2@PCM微胶囊的检测
采用日本日立公司的HITACHI S-4700型扫描电子显微镜(SEM)和日本电气公司的JEM-100CX-II型透射电子显微镜(TEM)对样品的形貌、结构、表面等特征进行观察.使用美国Thermo公司Nicolet 380傅里叶变换红外(FT-IR)光谱仪,通过添加溴化钾实现粉末样品压片,并最终得到样品的红外光谱曲线.使用美国Thermo Scientific公司的ANTARIS MX型傅里叶变换近红外(FT-NIR)光谱对样品的分子结构进行检测.将仪器的SabIR型漫反射探头固定在带有温度控制附件的固体粉末样品中,实时记录样品随温度变化的近红外光谱曲线.使用美国TA公司Q20差示扫描量热仪(DSC)测量相变材料的潜热/相转变温度.将少量样品置于标准的密封铝锅中,以固定的升降温速率对相变材料进行升降温循环实验.
3 结果与讨论
3.1 PMMA-SiO2@PCM微胶囊的显微结构分析
图1是以聚甲基丙烯酸甲酯-二氧化硅(PMMASiO2)有机/无机复合材料为壳层石蜡为芯材的PMMA-SiO2@PCM微胶囊相变材料的电子显微照片和粒度分布曲线.图1A是PMMA-SiO2@PCM微胶囊的SEM照片.微胶囊是形貌规则且粒度分布均匀的球形纳米材料,单分散粒径100 nm左右.微胶囊的核壳结构如图1B为负染的TEM照片.粒子外圈衬度较深的部分是胶囊的壳层,其内部的白色区域是芯材相变材料.微胶囊的平均粒径在200 nm左右(图1C).
3.2 DSC分析
图2是PMMA-SiO2@PCM微胶囊和石蜡的DSC对比曲线.如图2B所示,微胶囊升温过程的相变起始温度为17.81°C;降温过程的相变起始温度为20.74°C,其平均潜热(H)约28.34 J·g-1.与图2A相比较,微胶囊在降温过程的相变起始温度降低了1.66°C,其原因是由微胶囊的小尺寸效应引起的过冷现象.由相变过程平均潜热计算微胶囊的封装比率约为35%.这应是由小尺寸效应导致的过冷度引起的.
3.3 FT-IR和FT-NIR光谱分析
图3是PMMA-SiO2@PCM、PMMA@PCM和PCM的FT-IR光谱对比曲线.在PCM的光谱曲线中,2956、2921、2852和720 cm-1处的吸收峰分别是-CH3非对称伸缩峰、-CH2-非对称伸缩峰、-CH2-对称伸缩峰和摇摆振动及脱离相变材料平面的弯曲振动峰.相变材料在1465和1378 cm-1处的吸收峰是-CH2-弯曲振动峰.10在PMMA-SiO2@ PCM微胶囊的FT-IR曲线中,2956、2921、2852、720、1465和1378 cm-1处的吸收峰与相变材料的吸收峰一一对应,这可以进一步证明相变材料的存在.除此之外,C-H在2925 cm-1处的伸缩振动峰、酯类的C=O在1732 cm-1处的振动峰及C=C在1646 cm-1处的伸缩峰可以证明,胶囊中存在聚甲基丙烯酸甲酯.19在1456和3565 cm-1处的峰分别是酯群的C-H弯度振动和OH吸收峰.11,14,20,21将PMMA-SiO2@PCM的FT-IR曲线与PMMA@PCM微胶囊相比较,3565 cm-1处存在较强的吸收峰,这应是二氧化硅的添加导致羟基吸收峰强度增强的原因.
图4是PMMA-SiO2@PCM微胶囊与PMMASiO2的FT-NIR光谱对比曲线.PMMA-SiO2@PCM微胶囊在5671、5783和8250 cm-1处的吸收峰应是由相变材料吸收近红外光产生的结果.5380-5855 cm-1区域属于一级倍频振动带.22-24在5955 cm-1处的吸收峰是由C-CH3的二级非对称伸缩倍频引起的.除了在5123和8516 cm-1处的二级伸缩振动倍频外,其他峰是由组合频振动引起的.相变材料的近红外吸收峰特性:5671和5783 cm-1处的吸收峰是由PCM的-CH2的一级倍频引起的;8250 cm-1处的吸收峰是由-CH2的三级伸缩倍频引起的.1,25

图1 PMMA-SiO2@PCM材料的微结构图片及粒度分布Fig.1 Microstructure images and particle size distribution of PMMA-SiO2@PCM materials(A)SEM image;(B)TEM image;(C)particle size distribution

图2 DSC曲线分析Fig.2 DSC curve analyses(A)paraffin(PCM);(B)PMMA-SiO2@PCM

图3 PMMA-SiO2@PCM、PMMA@PCM和PCM的FT-IR曲线Fig.3 FT-IR curves of PMMA-SiO2@PCM, PMMA@PCM and PCM
3.4 PMMA-SiO2@PCM微胶囊相变材料在相变过程中的近红外光谱吸收特性
图5是PMMA-SiO2@PCM微胶囊熔化过程的FT-NIR光谱系列曲线.微胶囊从11.3°C开始加热至28.0°C.每0.5°C的温度间隔记录一次吸收曲线.样品放置在250 mL的烧杯中,使样品尽量薄地平铺于烧杯底部,以提高样品热传导的速度和受热均匀度.
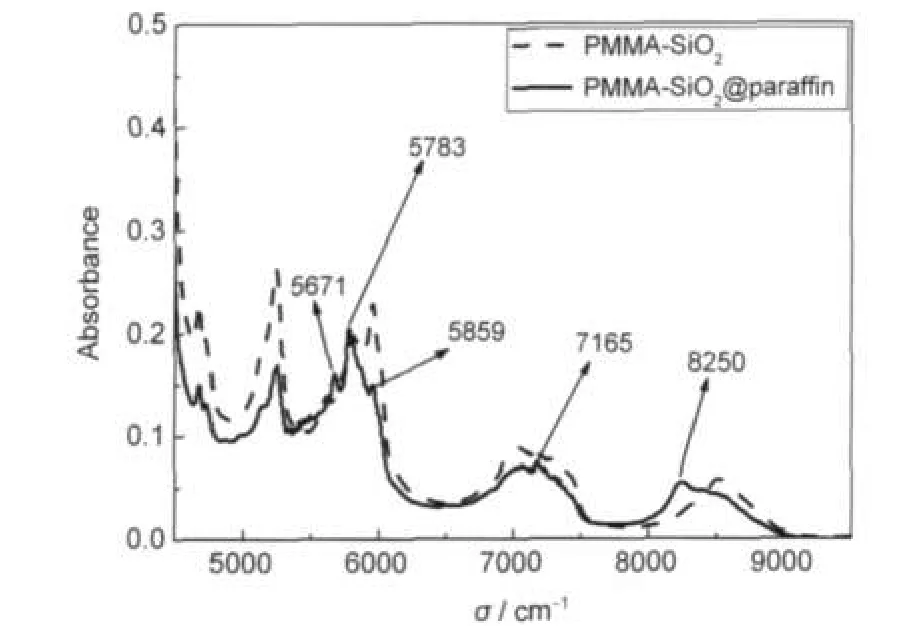
图4 PMMA-SiO2@PCM与PMMA-SiO2的FT-NIR光谱对比曲线Fig.4 Comparison of FT-NIR curves of PMMA-SiO2@PCM and PMMA-SiO2

图5 PMMA-SiO2@PCM微胶囊在熔化过程的FT-NIR光谱对比曲线Fig.5 Comparison of FT-NIR curves of PMMA-SiO2@PCM during melting processThe samples are heated from 11.3 to 28.0°C. The curves are recorded per 0.5°C.
在每个温度点处,实验采用任取样品5个位置求平均值的办法测量大量PMMA-SiO2@PCM微胶囊熔化过程的近红外光谱特性.图5的近红外曲线阵列中发现,在PMMA-SiO2@PCM微胶囊熔化过程中,4330、4440、5243、5780、7038和8250 cm-1处的六个强吸收峰在22°C左右峰的强度最大.六个强吸收峰在微胶囊熔化过程吸收峰强度的具体变化信息见图6.图中清晰可见,在22°C左右的温度条件下,微胶囊有最强吸收峰.温度继续升高,微胶囊的吸收峰强度突然降低.至28°C,吸收峰的强度变化波动不大.根据DSC分析结果,22°C左右是PMMASiO2@PCM微胶囊熔化速度最大点.
在每个温度点处,实验采用近红外探头位置固定的方法对PMMA-SiO2@PCM微胶囊熔化过程的FI-NIR变化情况进行分析检测.检测过程中,某一温度点检测5次求平均.检测结果见图7的PMMASiO2微球和图8的PMMA-SiO2@PCM微胶囊结构变化信息.图7显示,升温过程中,PMMA-SiO2微球的近红外吸收峰只在5241 cm-1处有较明显的振动变化.并且,随着检测温度的升高,吸收峰的强度逐渐增加至最大值.与之相比较,图8 PMMASiO2@PCM微胶囊有多个近红外吸收峰强度变化较明显.除8250 cm-1处之外,以4681 cm-1的C=O伸缩及-CH3对称伸缩一级倍频的组合频、4729 cm-1的C=O伸缩及-CH3非对称伸缩一级倍频的组合频、5243 cm-1的O-H伸缩及形变一级倍频组合频和7038 cm-1的-OH二级倍频及Si-O一级倍频的组合频引起的吸收峰强度变化最大.这些分子结构均来自PMMA-SiO2@PCM微胶囊的壳层,其吸收峰的强度随检测温度的增加而逐渐增大.来自相变材料的吸收峰的位置,以5671 cm-1的-CH2对称伸缩二级倍频、5783 cm-1的-CH2非对称伸缩二级倍频和5859 cm-1的-CH2对称伸缩二级倍频处的吸收峰有较强的强度变化,其他振动峰强度变化不明显.其中,5671和5859 cm-1的-CH2对称伸缩二级倍频吸收峰强度的变化要比5783 cm-1的-CH2非对称伸缩二级倍频吸收峰强度变化要强些.同时,对称伸缩吸收峰的强度变化是逐渐递增的,而非对称式伸缩振动峰的强度则是无规律的上下波动.根据上述分析推断,相变材料相变过程是-CH2对称伸缩振动和非对称伸缩振动共存的-CH2分子振动变化过程.随着温度的升高,-CH2对称伸缩振动变化较规律的逐渐增强.而对于-CH2非对称伸缩振动,则呈无规律振动状况.与来自壳层的吸收峰相比较,其强度的变化仅是壳层材料的吸收峰强度变化幅度的一半.
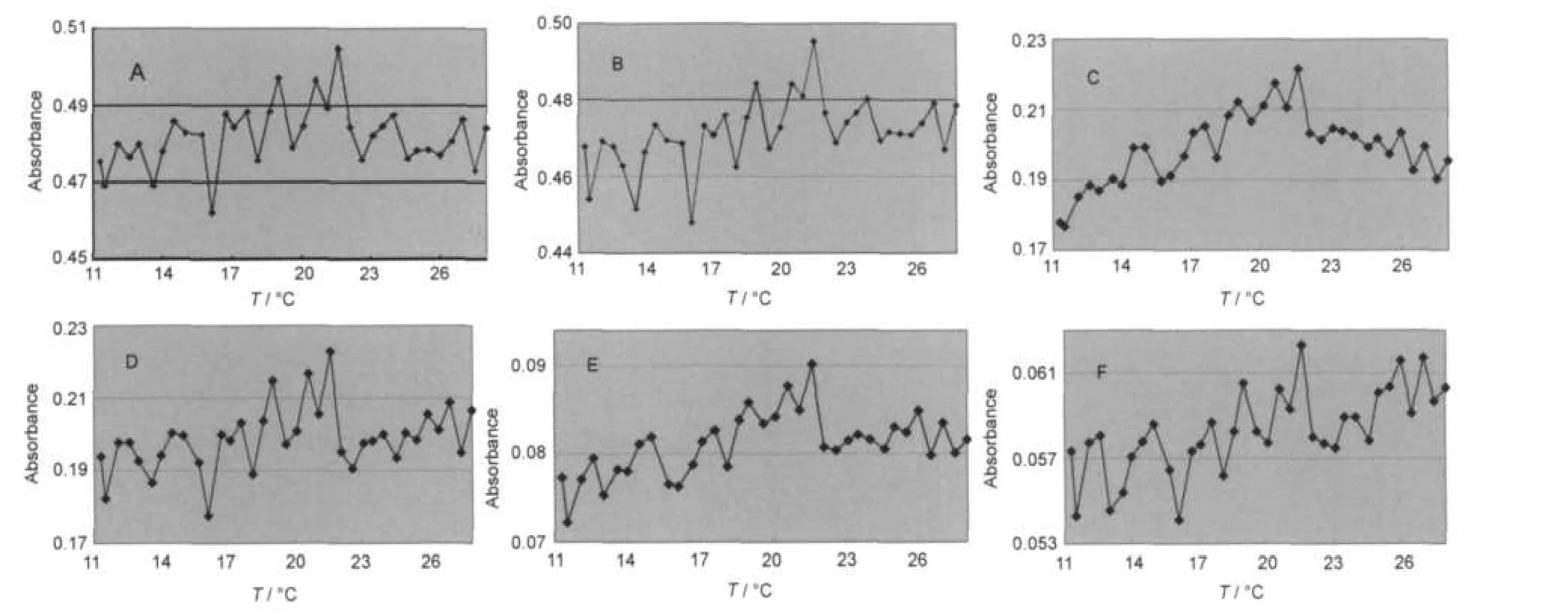
图6 熔化过程中PMMA-SiO2@PCM微胶囊的FT-NIR吸收峰强度变化对比曲线Fig.6 Comparison of FT-NIR absorbance of PMMA-SiO2@PCM during melting processσ/cm-1:(A)4330,(B)4440,(C)5243,(D)5780,(E)7038,(F)8250
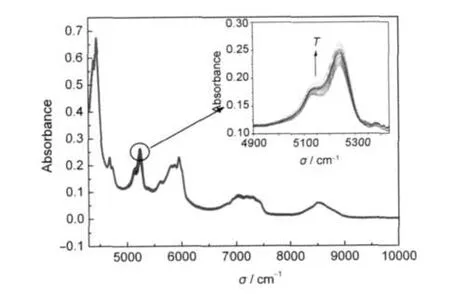
图7 PMMA-SiO2微球在升温过程的FT-NIR光谱对比曲线Fig.7 Comparison of FT-NIR curves of PMMA-SiO2 during heating processThe samples are heated from 11.0 to 28.1°C. The curves are recorded per 0.5°C.
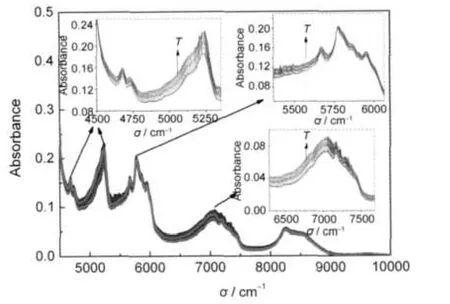
图8 PMMA-SiO2@PCM微胶囊在熔化过程的FT-NIR光谱对比曲线Fig.8 Comparation of FT-NIR curves of PMMA-SiO2@PCM during melting processThe samples are heated from 11.1 to 28.0°C. The curves are recorded per 0.5°C.
根据波尔兹曼分布,在室温条件下,受中红外光激发,分子以从基态到激发态的跃迁(n=0→n=1)为主.其他允许的激发态之间的跃迁,如n=1→n=2和n=2→n=3等,则受近红外激发而产生.虽然只有少量的分子处于激发态,而且由激发态跃迁引起的吸收峰强度较弱,但近红外光谱可以实现对材料结构变化的实时监测,而且可以对大量样品进行检测,提高检测的准确度.对于PMMA-SiO2@PCM微胶囊的体相检测结果而言,在22°C相变速度最大点之前,其强度逐渐增大的变化符合温度的升高有助于分子激发态跃迁的规律.1到22°C最大速度相变点,整个微胶囊的激发态跃迁都受到最大激发.22°C之后,相变材料的相变速率有一个突降.微胶囊体系趋向较平稳变化阶段.因此,在22°C之后,吸收峰强度突降,随后缓步振动变化.对于PMMA-SiO2@PCM微胶囊某一点处的分子振动情况分析结果而言,温度的变化对微胶囊结构的影响远超过对实心PMMA-SiO2微球结构的影响.温度的升高,使微胶囊壳层变形较大,整个分子体系较活跃,因此,处于激发态的分子数目增加,激发态之间跃迁的概率也相应地增加.所以,PMMA-SiO2@PCM微胶囊中壳层材料吸收峰强度随温度变化较大,且呈递增趋势.微胶囊中相变材料的吸收峰强度随温度的升高变化较小,这是因为相变材料相变过程温度保持不变,从而使处于激发态的分子数量增加较少,或不增加.
近红外光谱在线监测分析技术不但可以得到微胶囊相变材料壳材和芯材在相变过程的分子振动情况,同时,还可以用来辅助判断微胶囊的核壳结构.若制备的是核壳结构的微胶囊相变材料,则其壳层材料的大多数近红外光谱吸收峰都有较大的强度变化.否则,只有5241 cm-1处的吸收峰有较强的强度变化.
4 结论
通过FT-NIR光谱在线分析技术和DSC分析结果相结合的方式,实验得到了直径为100 nm的PMMA-SiO2@PCM相变机理的光谱分析结果及相关的微结构变化信息,微胶囊中,石蜡的融化过程就是-CH2分子对称伸缩振动逐渐增强和非对称伸缩无规则振动共存的振动变化过程.石蜡相变过程中,其近红外吸收峰强度的变化仅是壳层材料的吸收峰强度变化幅度的一半.即,PMMA-SiO2@PCM微胶囊熔化过程中,壳层随温度的升高而发生倍频跃迁的几率大于芯材相变材料.这是由于壳层较大的体积变化导致的跃迁几率的增加.同时,近红外光谱可以用来辅助分析微胶囊的核壳结构.核壳结构的微胶囊相变材料,其壳层材料的大多数近红外吸收峰在相变过程有较明显的强度变化.此外,近红外光谱可实现对微胶囊相变材料相变过程监测.在相变材料的最大相变点处,近红外光谱具有最强吸收峰.
(1) Lu,W.Z.Modern Near Infrared Spectroscopy Analytical Technology,2nd ed.;China Petrochemical Press:Beijing,2007; pp 16-345.[陆婉珍.现代近红外光谱分析技术(第二版).北京:中国石化出版社,2007:16-345.]
(2) Wurster,D.E.;Werawatganone,P.J.Pharm.Sci.2010,99, 1440.
(3) Green,B.K.;Lowell,S.Oil-Containing Microscopic Capsules and Method of Making Them.USAPat.Appl.2800457,1957.
(4) Rau,G.C.Science 1950,111,229.
(5) David,S.E.;Daniels,E.S.;El-Aasser,M.S.ACS Symposium Series 1992,492,1.
(6) Su,J.F.;Wang,L.X.;Ren,L.Colloid Polym.Sci.2005,284, 224.
(7)Wang,J.P.;Zhao,X.P;Guo,H.L.Langmuir 2004,20,10845.
(8) Frere,Y.;Danicher,L.;Gramain,P.Eur.Polym.J.1998,34,193.
(9) Kim,E.Y.;Do,K.H.J.Appl.Polym.Sci.2005,96,1596.
(10)Hawlader,M.N.A.;Uddin,M.S.;Khin,M.M.Appl.Energy 2003,74,195.
(11) Loxley,A.;Vincent,B.J.Colloid Interf.Sci.1998,208,49.
(12) Jorge,L.C.;Jeffrey,L.W.;Randolph,S.D.Langmuir 2008,24, 2064.
(13) Brinker,C.J.Advances in Chemistry 1994,234,361.
(14) Ogawa,K.;Chemburu,S.;Lopez,G.P.;Whitten,D.G.; Schanze,K.S.Langmuir 2007,23,4541.
(15) Joncheray,T.J.;Audebert,P.;Schwartz,E.;Jovanovic,A.V.; Ishaq,O.;Chávez,J.L.;Pansu,R.;Duran,R.S.Langmuir 2006,22,8684.
(16) Zhang,H.Z.;Wang,X.D.;Wu,D.Z.J.Colloid Interface Sci. 2010,343,246.
(17)Wu,X.L.;Wang,Y.H.;Sun,R.;Lai,M.B.;Du,R.X.;Zhang, Z.J.Journal of Physics-Conference Series 2009,188,012046.
(18) Mercedes,P.;Matias,J.;Mariana,B.M.;Susana,C.G.;Sara, A.B.Chem.Mater.2008,20,3015.
(19) Chang,C.C.;Tsai,Y.L.;Chiu,J.J.;Chen,H.Journal of Applied Polymer Science 2009,112,1850.
(20) Chang,K.C.;Chen,Y.K.;Chen,H.Journal of Applied Polymer Science 2008,107,1530.
(21) Zou,H.;Wu,S.S.;Shen,J.Chem.Rev.2008,108,3893.
(22) Reikichi,I.;Toshihiko,M.Anal.Chem.2007,79,3455.
(23) Matsushita,A.;Ren,Y.Z.;Matsukawa,K.;Inoue,H.;Minami, Y.;Noda,I.;Ozaki,Y.Vibrational Spectroscopy 2000,24,171.
(24) Chung,H.;Ku,M.S.Applied Spectroscopy 2003,57,545.
(25)Lu,W.Z.;Yuan,H.F.;Xu,G.T.;Qiang,D.M.Modern Near Infrared Spectroscopy Analytical Technology,1st ed.;China Petrochemical Press:Beijing,2000;pp 20-26.[陆婉珍,袁洪福,徐广通,强冬梅.现代近红外光谱分析技术(第一版).北京:中国石化出版社,2000:20-26.]
October 27,2010;Revised:February 11,2011;Published on Web:March 11,2011.*
.Email:rong.sun@siat.ac.cn;Tel:+86-755-86392158.
Near Infrared Spectrum Analysis of Organic/Inorganic Composite Microcapsule Phase Change Materials
WU Xiao-Lin1,2SUN Rong1,*ZHU Peng-Li1DU Ru-Xu3
(1Shenzhen Institutes of Advanced Technology,Chinese Academy of Sciences,Shenzhen 518055,Guandong Province,P.R.China;
2Graduate University of Chinese Academy of Sciences,Beijing 100049,P.R.China;3The Chinese University of Hong Kong,Hong Kong,P.R.China)
Using Fourier transform near infrared(FT-NIR)spectroscopy,the characteristics and phase change mechanism of the phase change process for PMMA-SiO2@PCM microcapsules were explored. We investigated changes in the microstructure of the microcapsules as well.The results show that the melting of paraffin molecules in the microcapsules is a process wherein the-CH2symmetric stretching vibration gradually increases and the asymmetric stretching vibration changes randomly.During the paraffin phase change process,the change in absorbance intensity is only half that of the shell materials. Additionally,NIR spectroscopy was used to analyze the core-shell structure and to monitor the phase change process for the microcapsules.The application of NIR spectroscopy to study the phase change process for microcapsule phase change materials has scientific significance and is of value to study the phase change mechanism for the determination of the most efficient phase change materials.
Microcapsule;Fourier transform near infrared;Phase change material; Phase change;Organic/inorganic composite material
O641
The project was supported by the National Natural Science Foundation of China(20971089).国家自然科学基金(20971089)资助项目
猜你喜欢
北京航空航天大学学报(2022年8期)2022-08-31
食品安全导刊(2021年21期)2021-08-30
空间科学学报(2021年1期)2021-05-22
雷达学报(2018年1期)2018-04-04
光学精密工程(2016年2期)2016-11-07
中国光学(2015年5期)2015-12-09
中国光学(2015年5期)2015-12-09
中国塑料(2015年6期)2015-11-13
中国塑料(2015年9期)2015-10-14
橡胶工业(2015年4期)2015-07-29
